












【佳学基因检测】基因检测如何从外周血细胞得知乳腺癌的存在?病例与健康对照大人群研究
外周血细胞乳腺癌基因检测导读:
肿瘤与宿主的相互作用超出了局部微环境,癌症的发展在很大程度上取决于恶性细胞劫持和利用宿主正常生理过程的能力。在这里,乳腺癌基因检测技术领选策略保证团队确定当存在未经治疗的乳腺癌 (乳腺癌) 时,外周血细胞内的许多基因表现出差异表达,并利用这一事实构建了一个 50 基因特征,将 乳腺癌 患者与基于人群的对照区分开来。乳腺癌基因检测技术领选策略保证团队的结果来自乳腺癌基因检测技术领选策略保证团队独特的基于人群的挪威妇女和癌症队列中的一系列大型数据集,这些数据集使乳腺癌基因检测技术领选策略保证团队能够研究药物和肿瘤特征对乳腺癌基因检测技术领选策略保证团队基于血液的测试的影响,并在两个外部数据集中进行了进一步测试。乳腺癌基因检测技术领选策略保证团队的 50 基因特征包含细胞抑制信号,包括对免疫反应的特异性抑制,以及影响这些过程中转录的药物被确定为混杂因素。通过分析血液中差异表达的生物学过程,乳腺癌基因检测技术领选策略保证团队能够解释为什么宿主的全身反应可能是 乳腺癌 的可靠标志物,其特征是免疫特异性途径和“通用”细胞的低表达由 MYC 驱动的程序(即,新陈代谢、生长和细胞周期)。总之,外周血细胞的基因表达受到乳腺癌特异性存在的显着干扰,这些变化同时涉及许多与 乳腺癌 免疫逃逸相关的系统性细胞抑制信号。
基因检测如何从外周血细胞得知乳腺癌的存在课题创新点?
血细胞是信息的动态仓库。就癌症而言,研究表明血细胞具有与实体瘤相关的遗传特征。在本研究中,发现外周血细胞中的基因在未经治疗的乳腺癌女性中存在差异表达,从而开发出能够识别患有该疾病的女性的 50 个基因特征。基因特征包括免疫抑制特异性信号。乳腺癌与免疫特异性通路的低表达以及与 MYC 驱动的“通用”细胞程序的关联可以解释宿主的全身反应。
关键词: 乳腺癌,基于人群的病例对照研究,肿瘤-宿主相互作用,血液基因表达谱
癌症不仅仅是自主的细胞群;它们分泌可引起宿主全身反应的可溶性因子,而宿主反应反过来又会影响癌细胞。几项研究支持这样的观点,即肿瘤系统地作用以调节整体癌症进展并且癌症发展在很大程度上取决于恶性细胞劫持和利用宿主正常生理过程的能力。尽管人们对癌症中癌症与宿主相互作用的认识越来越多,但乳腺癌基因检测技术领选策略保证团队对宿主如何响应癌症信号并进而影响肿瘤进展和预后的理解还很初步。越来越多的证据表明,外周血细胞的基因表达谱是评估与实体瘤相关的基因特征的有价值的工具。包括乳腺癌基因检测技术领选策略保证团队在内的几个小组已经定义了健康个体中血液基因表达的个体内部和个体间变异性,并建立了血液样本采集和基因表达谱分析的标准化程序。在这项研究中,乳腺癌基因检测技术领选策略保证团队在挪威妇女和癌症研究 (NOWAC) 中选择了大量的乳腺癌 (BC) 患者和代表一般人群的女性,这些患者的出生年份和随访时间相匹配。据乳腺癌基因检测技术领选策略保证团队所知,这是该领域的第一项研究,准确地代表了从代表一般人群的女性中识别乳腺癌患者的实际情况。乳腺癌基因检测技术领选策略保证团队遵循严格的实验设计,在从 RNA 扩增到杂交的实验室程序的所有步骤中,每个病例样本都使用年龄匹配的对照进行处理。在两个训练集和一个验证集中进行了配对分析,以消除任何技术偏差并帮助确保结果的普遍性。首先根据生活方式暴露和肿瘤特征评估已识别的 50 基因血液特征的准确性。然后使用来自乳腺癌患者的血细胞或分离的免疫细胞谱在两个额外的外部基因表达数据集中进一步测试该特征,乳房 X 光检查可疑或诊断患有良性乳腺疾病的女性。乳腺癌基因检测技术领选策略保证团队的多基因特征的行为也在其他癌症类型中进行了评估,以评估系统对乳腺癌的特异性。最后,乳腺癌基因检测技术领选策略保证团队研究了宿主全身反应所涉及的分子过程,并提供了为什么基于血液的基因表达可能含有乳腺癌存在的可靠信号的基本原理。
基因检测如何从外周血细胞得知乳腺癌的存在研究方法
NOWAC 研究
NOWAC 研究由 1991 年至 2006 年招募的 172,471 名 30 至 70 岁的女性组成,她们回答了一到三份关于饮食、药物使用和生活方式的问卷。十家挪威最大的医院参与从乳腺癌事件中采集血液和肿瘤组织。与挪威乳腺癌小组合作,参与 NOWAC 研究的 1943 年至 1957 年间出生的每位女性被要求在合作医院接受诊断活检或接受乳腺癌手术,在手术和治疗前捐赠肿瘤活检和两份血样,一份收集到 PAXgene™ 管(PreAnalytiX GmbH,Hembrechtikon,瑞士)中用于基因表达分析,另一份收集到柠檬酸盐管中。参与者还被要求回答一份两页的问卷,主要收集关于当前使用激素和药物、饮酒和吸烟习惯的信息。然后将生物样本在-70°C 的特罗姆瑟隔夜邮寄用于生物库。同时,为每个乳腺癌病例接近五个对照,以便从每个病例至少两个对照获得血样。对照是随机抽取的,但与纳入 NOWAC 队列的时间和出生年份相匹配。人体生物材料已获得挪威地区医学和健康研究伦理委员会的批准,并符合挪威的生物样本库法律。
2009 年,从后基因组生物库 (CC1) 中选择了来自病例的 96 个血液样本和每个病例的两个匹配对照。2010 年,从后基因组生物库 (CC2) 中选择了从病例采血后 4 天内收到的 63 份血液样本和每个病例的匹配对照。2011 年,从后基因组生物库 (CC3) 中选择了从病例采血后 4 天内收到的 90 份血液样本和每个病例的匹配对照。
微阵列数据采集
为了控制技术变异性,例如试剂和试剂盒的不同批次变化、日常变化、微阵列生产批次和与不同实验室操作员相关的影响,每个案例通过 RNA 提取与一个相应的匹配对照分组(CC1 中的 RNA 提取除外)随机运行)、扩增和杂交。根据挪威特隆赫姆 NTNU Genomics Core Facility 的制造商手册,使用 PAXgene Blood miRNA Isolation Kit 从病例和每个病例的匹配对照中分离总 RNA。分别使用 NanoDrop ND-8000 分光光度计(ThermoFisher Scientific,Wilmington,Delaware)和安捷伦生物分析仪(Palo Alto,CA)评估 RNA 数量和纯度。使用 300 ng 总 RNA 和 Illumina® TotalPrep™-96 RNA 扩增试剂盒(Ambion, Austin, TX)在 96 个板中进行 RNA 扩增。CC1 和 CC2 中包含的病例和对照在 IlluminaHumanAWG-6 第 3 版表达珠芯片上运行。CC3 中包含的病例和对照在 IlluminaHumanHT-12 第 4 版表达珠芯片上运行。来自 Illumina (San Diego, CA) 的 GenomeStudio 用于评估每个阵列的质量。
微阵列数据预处理
使用 R ( http://cran.r-project.org ) 和来自 Bioconductor 项目 ( http://www.bioconductor.org ) 的工具进行微阵列数据预处理和分析,以适应乳腺癌基因检测技术领选策略保证团队的需要。
对所有三个独立数据集进行相同的数据预处理(图1)。乳腺癌基因检测技术领选策略保证团队排除了收集后超过 4 天收到的样本和 RNA 质量低 (RIN < 7) 的样本。从癌症登记处更新后发现误诊的样本或 lumiR 软件包调用的异常值,对数据集进行了修剪。更准确地说,当离群中心的欧几里德距离大于到中心的中值距离的两倍时,就可以识别异常值。聚类中心定义为去除离中心最远的 10% 样本后所有样本的平均值。最后,从上述排除程序中得到的不匹配样本被排除在分析之外。使用 lumiR 包分别在每个数据集中对微阵列数据进行预处理。如果一个探针的强度与背景强度显着不同(p值 < 0.05),从而分析了 CC1 和 CC2 中的 48,803 个探针,以及 CC3 中的 47,323 个探针。乳腺癌基因检测技术领选策略保证团队排除了在至少 70% 的样本中没有表达值的所有探针,分别导致 CC1、CC2 和 CC3 中的 13,460、10,341 和 12,519 个探针。执行方差稳定并通过分位数归一化对数据进行归一化。乳腺癌基因检测技术领选策略保证团队对 Illumina 阵列第 3 版用于 CC1 和 CC2 以及第 4 版用于 CC3 使用了重新注释管道。具有相似基因符号的探针强度平均导致 CC1、CC2 和 CC3 中分别有 9,338、7,898 和 8,529 个独特的基因符号。微阵列数据已保存在欧洲基因组表型档案 (EGA; https://www.ebi.ac.uk/ega/ ) 登录号 EGAS00000000134。
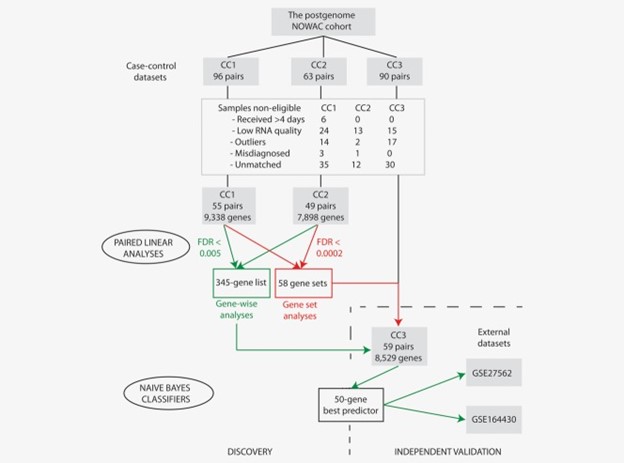
图1:课题研究流程图。显示了来自挪威妇女和癌症 (NOWAC) 研究 (CC1、CC2 和 CC3) 的三个独立病例对照数据集的样本排除。进行配对线性分析以鉴定单个基因(错误发现率,FDR < 0.005)和基因组(FDR < 0.002)在乳腺癌病例对照对中差异表达。使用朴素贝叶斯分类器基于在 CC1 和 CC2 中差异表达的 345 个基因的表达预测 CC3 中乳腺癌的存在。在 345 个基因列表中进一步选择了 50 个基因,并在来自 NCBI 基因表达综合数据库的两个外部数据集中进行验证(包括来自乳腺癌患者、良性乳腺疾病患者、对照、胃肠道和大脑的外周血单个核细胞 (PBMC) 的基因表达谱)癌症患者 ( GSE27562 ) 和来自乳腺癌患者和对照组的外周血细胞的基因表达谱以及疑似乳房 X 线照片 ( GSE164430 )。[颜色图可以在在线问题中查看,该问题可在wileyonlinelibrary.com 获得。]
技术可变性
扩增日期与乳腺癌基因检测技术领选策略保证团队的基因表达谱相关,但后者仍然独立于乳腺癌状态,因为每个病例都在同一轮中用其对照进行扩增,并进行了配对分析。在CC1中,在CC1中随机抽取血液样本进行RNA提取,并且RNA提取日期与疾病状态显着相关(χ 2检验:p值= 0.02)。支持信息 附加文件 1 显示了基于 CC1 中大多数可变基因的前五分位数表达的样本排序。总体而言,六个不同 RNA 提取日期的变量编码似乎与血液基因表达谱没有密切关联,因此在进一步的分析中尚未调整其影响。
基因配对线性分析
为了识别在病例和对照之间差异表达的单个基因,乳腺癌基因检测技术领选策略保证团队使用在每个数据集中的软件包 Limma 中实施的经验贝叶斯方法进行了配对基因线性分析。计算错误发现率 (FDR) 以针对多次测试进行调整。
预测乳腺癌的存在
尽管独立性假设是不准确的,朴素贝叶斯算法缓解了源自维数灾难的问题,并被实施为类预测方法。事实上,朴素贝叶斯是一种用于基因表达研究的成熟学习方法,因为它简单、可解释,并且已被证明具有与更复杂的方法相似甚至有时超过的性能。在存在乳腺癌的情况下,在 CC1 和 CC2 中通常差异表达的基因中进行基因选择(配对线性分析 FDR < 0.005;N = 345,图1) 以优化乳腺癌基因检测技术领选策略保证团队跨数据集签名的稳健性。首先使用 CC3 ( N = 341)中表达的所有基因构建一个朴素贝叶斯分类器 (NBC), 并通过留一法交叉验证 (LOOCV )进行测试。预测准确性是真实与所有预测都来自 NBC 生成的后验概率。NBC 的重要性通过 Fisher 精确检验来测试,该检验测量观察到的和预测的疾病状态之间的关联强度。然后将乳腺癌基因检测技术领选策略保证团队的分类器显着性与从 100,000 个 NBC 获得的显着性背景分布进行比较,其中包括从 CC3 中表达的 8,529 个基因中随机选择的 341 个基因。同样,乳腺癌基因检测技术领选策略保证团队构建了包括几个子集大小(10、25 或 50 个基因)的 341 个重叠基因在 CC3 中表达的 NBC,其中大小为 50 的预测因子似乎最合适(支持信息附加文件 4A)。乳腺癌基因检测技术领选策略保证团队还测试了 100,000 个大小为 50 的 NBC,从更大的基因列表中随机选择,这些基因通常因乳腺癌的存在而在 CC1 和 CC2 中差异表达(N = 565 个 FDR < 0.01 的基因和N = 1,426 个 FDR < 0.05 的基因),但在预测变量显着性方面没有任何改善(支持信息附加文件 4B)。“最佳” 50 基因预测因子是根据其在预测乳腺癌存在方面的统计显着性(Fisher 检验),在使用 CC3 中表达的 341 个基因中的 50 个基因构建的 100,000 个预测因子中凭经验选择的。其重要性进一步与 100,000 个随机 50 基因 NBC 的背景分布进行了比较。
在所有三个数据集中,乳腺癌基因检测技术领选策略保证团队使用学生或卡方检验调查了由 RIN 值、个体或暴露变量(如年龄、BMI 或吸烟状况)量化的 RNA 质量,以及使用更年期激素疗法或其他特定药物是否可以解释控制(即假阳性)和案例(即.,假阴性)。以同样的方式,乳腺癌基因检测技术领选策略保证团队调查了肿瘤受体状态(雌激素和孕激素受体)或阶段是否与乳腺癌基因检测技术领选策略保证团队在所有三个数据集中的 50 基因预测因子错误分类的病例相关。除了 CC3 中对照组使用特定药物外,这些变量均与对照组或病例的错误分类无关(见结果)。值得注意的是,BC 主要是 ER 阳性和 I 期或 II 期。 最后,乳腺癌基因检测技术领选策略保证团队从连接图中研究了扰动特征 ( n > 10),显着丰富了乳腺癌基因检测技术领选策略保证团队的 50 基因预测因子。
使用 NCBI 的基因表达综合中存放的两个额外外部数据集对“最佳”50 基因预测因子进行独立验证,包括来自乳腺癌患者、良性乳腺疾病患者、对照、胃肠道患者的外周血单个核细胞 (PBMC)的基因表达谱和脑癌患者 ( GSE27562 ) 以及来自乳腺癌患者和对照组的全血细胞的基因表达谱以及可疑乳房 X 线照片 ( GSE164430 ;图1)。乳腺癌基因检测技术领选策略保证团队用雨果基因命名委员会指定的符号表示基因。
功能聚类和通路分析
与乳腺癌诊断相关的基因列表的功能聚类使用位于http://david.abcc.ncifcrf.gov/的注释、可视化和集成发现数据库 (DAVID)进行。对于每个功能簇,乳腺癌基因检测技术领选策略保证团队选择了 FDR < 0.1 的项,并计算了中位倍数富集和 FDR。
基因组分析
使用 GSVA 计算每个样本的分子特征数据库的 C2(策划基因集)和 C5(基因本体基因集)子集的 3.0 版中包含的通路(大小;最小 = 5 和最大 = 500)的富集分数R 包。乳腺癌基因检测技术领选策略保证团队使用不超过三种免疫细胞亚型的 CD 标记和在每个分化的免疫细胞亚型中特异性过表达的转录物构建特定于免疫细胞亚型的基因集,并以相同的方式计算每个样本的富集分数。如前所述,乳腺癌基因检测技术领选策略保证团队使用配对线性分析测试了病例对照对的富集分数之间是否存在差异。
通过全局测试协变量分析估计感兴趣通路内的每个基因或样品对差异表达的总体测试统计的显着贡献。globaltest R 包中的协变量和主题图估计每个(集群)基因(协变量)或样本(主题)对差异表达的总体测试统计数据的贡献,绘制p替代的单个组件的测试值。样品按降序排列。使用相关性作为距离度量,在层次聚类中对基因进行排序。层次聚类图在作为聚类图顶部的完整集和作为叶子的单个协变量之间引入了测试协变量的子集。所有 2 k - 1 个集合上的继承过程 控制全族错误率,同时考虑到图的结构。
基因检测如何从外周血细胞得知乳腺癌的存在研究结果
BC的全血转录信号及其检测BC的潜力
疾病状态与包括在 CC1 中的病例对照对之间血液基因表达谱的显着差异相关(p = 6 × 10 -8,全局检验)。BC 的这种全血信号通过基于最可变基因表达的疾病状态对样本进行分组来说明(支持信息附加文件 2)。乳腺癌基因检测技术领选策略保证团队确定了 3,479 个基因在乳腺癌病例和对照对中表现出显着的表达差异(FDR < 0.005;配对线性分析),倍数变化的中值绝对值相对较低,等于 1.13。这表明与乳腺癌相关的基因表达变化幅度相对较低,但在外周血细胞中一致且普遍存在。
为了测试这些发现在独立数据集 (CC2) 中是否可复制,乳腺癌基因检测技术领选策略保证团队调查了另外 49 对乳腺癌病例和对照的血液基因表达谱(图1)。总共通过质量控制的 7,898 个基因中有 418 个在 CC2 中差异表达,FDR < 0.005,其中 345 个在 CC1 中也有差异表达(p = 3 × 10 -60,超几何检验;图 2a , 支持信息附加文件 3)。值得注意的是,BC 病例和所有 345 个重叠基因的对照之间差异表达的方向性在数据集之间是保守的(图 2b)。当根据 345 个重叠基因的表达总和对患者进行排序时,来自乳腺癌病例的大部分血液样本在两个数据集中与对照分离(图 2c)。
 患者外周血细胞的基因表达变化.jpg)
图 2:与对照组相比,乳腺癌 (BC) 患者外周血细胞的基因表达变化。( a ) 维恩图描绘了与初级 (CC1) 和次级 (CC2) 数据集中的对照相比,BC 患者外周血细胞中差异表达的基因之间的重叠。通过 CC1 和 CC2 中的配对线性分析,在 FDR < 0.005 时评估差异表达。使用超几何检验计算两个基因列表之间重叠的显着性。(b)在 CC1 和 CC2 中差异表达的 345 个重叠基因的表达倍数变化。CC1 中的对数倍数变化 (log FC) 绘制在 x 轴上,相对于y上 CC2 中相同基因的 log FCs-轴。两个数据集中存在 BC,绿色的基因表达不足。红色的基因在两个数据集中都因乳腺癌的存在而过度表达。(c)根据这 345 个重叠基因的表达总和,对乳腺癌病例(红色)和对照(粉红色)的血液样本进行排序。热图颜色代表对数空间中以均值为中心的倍数变化表达式。( d ) 朴素贝叶斯分类器在使用 Fisher 检验计算的验证数据集 (CC3) 中的重要性。垂直绿线表示基于 345 个重叠基因表达的朴素贝叶斯预测因子的重要性。绿色虚线表示可以从使用 CC3 中存在的 345 个随机基因构建的 100,000 个朴素贝叶斯预测变量中获得的显着性分布(N = 8,529)。纯红线表示可从 100,000 个预测因子中获得的显着性分布,这些预测因子使用 345 个重叠基因的 CC3 中表达的 341 个基因中的 50 个基因构建。红色虚线表示可以从使用数据集中存在的 50 个随机基因构建的 100,000 个预测变量中获得的显着性分布 ( N = 8,529)。
使用 CC1 和 CC2 选择与对照组相比在乳腺癌患者血细胞中差异表达的基因(N = 345,支持信息附加文件 3),乳腺癌基因检测技术领选策略保证团队使用 CC3 中表达的 341 个基因构建了一个预测因子(CC3 中不存在四个基因) 并在此验证数据集中准确预测疾病状态 ( p = 8.7 × 10 -5;Fisher 检验;图 2d )。值得注意的是,来自 CC3 中血液样本的扩增 RNA 使用不同版本的 Illumina 阵列系统进行杂交。“最好的” 50 基因预测因子(图 2d,支持信息附加文件 3) 在 341 个重要基因中选择的准确度为 72.9%,以预测验证数据集中存在 BC(敏感性 = 83.1% 和特异性 = 62.7%;p = 3.0 × 10 -9;Fisher's测试)。值得注意的是,来自连接图的基因表达特征26与组蛋白去乙酰化酶、热休克蛋白90、酪氨酸激酶和免疫反应抑制剂相关,乳腺癌基因检测技术领选策略保证团队的 50 基因预测因子阳性富集(支持信息附加文件 5)。很大一部分被错误分类为 CC3 病例的对照组(36.4%)目前正在使用选择性 5-羟色胺再摄取抑制剂(ATC N06AB)或选择性 β 受体阻滞剂(ATC C07AB)。先前发现与 CC3 中错误分类的对照相关的两种药物都能抑制 T 细胞和适应性免疫相关基因的表达。这可以解释乳腺癌基因检测技术领选策略保证团队的 50 基因预测因子在 CC3 中的较低特异性。总体而言,这表明乳腺癌基因检测技术领选策略保证团队的 50 基因预测因子包含细胞抑制信号,包括特异性抑制免疫,并且影响这些过程中涉及的转录的药物可能是与乳腺癌存在相关的基于血液的信号的混杂因素。
为了进一步验证结果,乳腺癌基因检测技术领选策略保证团队调查了乳腺癌基因检测技术领选策略保证团队是否能够在 NCBI 的 Gene Expression Omnibus 中存放的两个外部数据集中预测乳腺癌诊断,包括来自乳腺癌患者、良性乳腺疾病患者、对照、胃肠道和脑癌患者的 PBMC的基因表达谱图 ( GSE27562 ),以及来自乳腺癌患者和对照可疑乳房 X 线照片的外周血细胞的基因表达谱 ( GSE164430 ;图1)。在 PBMC 数据集中,与对照相比,乳腺癌基因检测技术领选策略保证团队的 50 基因预测器能够准确预测乳腺癌的存在(91.5% 准确度,支持信息附加文件 6)。这表明乳腺癌基因检测技术领选策略保证团队从外周血细胞中鉴定出的乳腺癌诊断特征存在于分离的免疫细胞中,包括单核细胞、T 细胞、B 细胞和自然杀伤 (NK) 细胞。来自其他癌症类型的所有 PBMC 样本均未预测为 BC,这表明乳腺癌基因检测技术领选策略保证团队的预测因子对乳房癌具有特异性。由于乳腺癌基因检测技术领选策略保证团队的预测器没有经过训练来区分恶性乳腺癌和良性乳腺疾病,当乳腺癌基因检测技术领选策略保证团队包含这些样本时,乳腺癌基因检测技术领选策略保证团队获得的准确度显着降低(63.4% 准确度;支持信息附加文件 6)。乳腺癌基因检测技术领选策略保证团队的 50 个基因预测因子中只有 33 个基因的表达在第二个数据集中可用(GSE164430)虽然乳腺癌基因检测技术领选策略保证团队能够显着预测乳腺癌诊断与怀疑筛查乳房 X 线照片的女性相比(p = 0.008;Fisher 检验,支持信息附加文件 7)。总之,乳腺癌基因检测技术领选策略保证团队的基于血液的基因表达分析在微阵列平台和外部数据集上产生了独特的稳健和可重复的结果,以从基于人群的对照中特异性检测 BC,需要进一步分析因外周血细胞中存在乳腺癌而发现失调的过程包括 PBMC。
通路和基因组分析
功能聚类表明,乳腺癌基因检测技术领选策略保证团队的 345 个基因列表丰富了与细胞凋亡、RNA 结合、剪接体/RNA 剪接、蛋白质合成、RNA 代谢、转录调控、细胞周期、代谢和信号转导相关的基因本体类别(表1)。专家策划的功能注释揭示了参与免疫过程、细胞生长/增殖、细胞骨架调节以及蛋白质和细胞代谢的额外基因组(支持信息附加文件 3)。
表1:与 345 个基因列表相关的显着富集的功能注释聚类(错误发现率,FDR < 0.10)在乳腺癌病例对照对中差异表达
|
注释项,N |
注释集群(关键字) |
基因,1 N |
倍数增加2 |
FDR2 (%) |
|
8 |
细胞死亡、凋亡 |
35 |
2.7 |
0.01 |
|
11 |
细胞凋亡的调节 |
37 |
2.3 |
0.01 |
|
5 |
RNA分解代谢过程 |
9 |
10.4 |
0.07 |
|
15 |
RNA结合、蛋白质合成、翻译、核糖体 |
54 |
3.9 |
0.05 |
|
2 |
RNA结合蛋白,RRM |
35 |
3.7 |
3.80 |
|
11 |
RNA加工、剪接、剪接体 |
29 |
3.1 |
1.73 |
|
3 |
蛋白激酶结合,酶结合 |
23 |
3.2 |
3.27 |
|
6 |
蛋白磷酸酶活性,锰 |
11 |
5.0 |
2.28 |
|
2 |
对无机物或金属离子的反应 |
12 |
3.2 |
6.02 |
|
2 |
核糖体,核糖核蛋白生物发生 |
11 |
3.6 |
3.48 |
|
7 |
转录调控、大分子代谢过程、含氮化合物、RNA pol II |
41 |
3.7 |
2.53 |
|
3 |
核苷酸或 ATP 结合 |
67 |
1.6 |
6.82 |
|
1 |
乙酰化氨基端 |
23 |
4.6 |
2.44 |
|
1 |
内质网 |
23 |
1.8 |
9.00 |
|
2 |
细胞周期 |
32 |
2.1 |
0.53 |
|
1 |
十四烷基化 |
5 |
7.4 |
6.06 |
|
2 |
前体代谢物和能量的产生,糖酵解 |
16 |
6.0 |
5.25 |
|
1 |
氧化还原酶活性,作用于供体的硫基团 |
5 |
6.7 |
8.72 |
|
1 |
Ras蛋白信号转导 |
8 |
3.8 |
8.42 |
1 345 个基因列表中参与相应过程的基因数量。
2集群中包含的所有重要注释术语的中值。
为了进一步研究乳腺癌如何影响血液中的基因表达,乳腺癌基因检测技术领选策略保证团队在 CC1 和 CC2 数据集中进行了基因集变异分析 (GSVA),并在 CC3 中验证了结果(图1)。乳腺癌基因检测技术领选策略保证团队发现 58 个基因组在 CC1 和 CC2 的病例对照对差异表达的前 200 个基因组之间重叠(FDR < 2 × 10 -4)。虽然乳腺癌基因检测技术领选策略保证团队之前在 CC3 中发现了一个与当前对照组使用特定药物的混杂因素,但在 CC1 和 CC2 中重叠的 58 个重要基因组中,有 45 个在 CC3 中得到验证(FDR < 0.15),并且根据疾病显示出显着可比的富集分数所有三个数据集中的状态(图 3)。GSVA 揭示了在对乳腺癌基因检测技术领选策略保证团队的 345 个基因列表进行功能聚类后看到的类似过程,包括转录和细胞骨架调节、细胞周期、细胞凋亡和代谢途径,但还发现了特别涉及抗原加工和呈递 (APP) 和 MYC 靶基因的其他基因特征(图 3)。
和对照(粉红色.jpg)
图 3:根据乳腺癌病例(红色)和对照(粉红色)在初级 (CC1)、二级 (CC2) 和验证 (CC3) 病例对照中差异表达的 45 个显着基因集的富集评分对血液样本进行排序系列。热图颜色代表对数空间中以平均为中心的折叠变化富集分数。
BC 和宿主的免疫系统
BC患者血细胞中APP通路表达减少是BC存在影响外周免疫效应细胞的最直接证据(图 3)。乳腺癌基因检测技术领选策略保证团队研究了重叠的核心基因(多重校正p值 <0.1),这些基因驱动观察到的 APP 通路与 CC1 和 CC2 数据集中存在的乳腺癌的关联(图 4a )。与对照组相比,作为 MHC II 类途径一部分的所有核心基因在乳腺癌患者的血液样本中均表达不足,包括干扰素 γ 诱导蛋白 30 和参与 MHC II 类限制性表位的内吞产生的组织蛋白酶 S 以及CD74参与 MHC II 类蛋白的形成和运输,CD4是辅助 T 细胞受体 (TCR) 的辅助受体。在 MHC I 类通路中,免疫蛋白酶体的PSME3被定义为乳腺癌患者中低表达的 APP 通路中的核心基因。此外,编码蛋白酶体的三个基因(PSMB2、PSMB10和PSMD1)在乳腺癌基因检测技术领选策略保证团队的 345 基因列表(支持信息附加文件 3)中,与对照组相比,BC 患者的血细胞中表达不足。此外,直接参与肽加载到 MHC I 类分子(TAPBP和CALR)上的核心基因和编码 TAP 上游热休克蛋白 70 的基因在乳腺癌患者中表达不足。
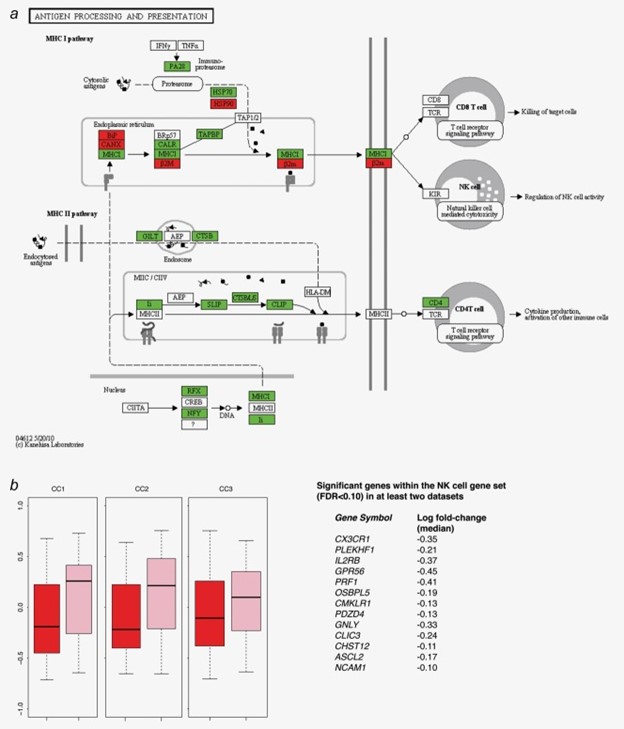
图 4:抗原加工和呈递途径 (APP) 和自然杀伤 (NK) 细胞基因集的基因集变异分析。( a ) 来自 KEGG 的 APP。驱动所观察到的 APP 与 CC1 和 CC2 中存在的乳腺癌关联的重叠核心基因根据它们在乳腺癌患者中的过度(红色)或不足(绿色)表达而着色。(乙) 箱线图表示来自 NK 细胞基因集的基因集变异分析的富集分数,该基因集与来自 CC1、CC2 和 CC3(左)中的乳腺癌患者(红色)和对照(粉红色)的基因表达谱相关。NK 基因集中包含的基因列表与配对线性分析中的疾病状态显着相关,三个数据集中的至少两个数据集中的 FDR < 0.10 及其在所有数据集中的相应中值倍数变化(右)。
最后,与对照组相比,NK 细胞特异性基因在该组和来自乳腺癌患者的样本中始终低表达(图 4b)。与这一发现一致,与 CC1 和 CC2 中的对照相比,来自乳腺癌患者的血液样本中来自 KEGG 的 NK 细胞介导的细胞毒性途径显着低表达(平均 FDR = 0.004;配对线性分析)。
BC 患者外周血细胞中 Myc 的低表达和代谢、生长和增殖降低
根据 Myc Target Gene Database ,乳腺癌基因检测技术领选策略保证团队发现在乳腺癌患者的血细胞中由 Myc 调节的靶基因集表达减少(图 3)。被定义为核心基因的 Myc 靶标参与了对细胞生长和增殖具有特定影响的全球基因调控网络(ILK、ARPC4、PP2R4、ERBB2和CEBPA)。
比较各组之间的总 RNA 水平,以研究 Myc 最近被重新构建为所有活性基因的基因表达的一般放大器的效果。来自乳腺癌病例的38 个样本的 RNA 水平低于来自匹配对照的血液样本(p值 = 8 × 10 -6;逻辑回归)。仅通过 RIN 值测量的 RNA 降解不能解释与对照相比,BC 患者血液样本中 RNA 产量的降低(数据未显示)。在进一步尝试提取乳腺癌患者外周血细胞中 Myc 作用的基本要素时,强调了另一组被接受为真正 Myc 靶标的基因之间的结合和表达变化。乳腺癌基因检测技术领选策略保证团队比较了前五分之一样本中的 RNA 浓度和MYC的表达,这对真正的 Myc 靶基因集的富集意义贡献最大(支持信息附加文件 11)。MYC的表达与 RNA 浓度显着相关,与对照组相比,BC 患者血液样本中MYC的显着低表达(图 5)。与此一致,大多数差异表达的转录因子,与确认稳态转录率降低的对照相比,作为一般转录机制一部分并参与染色质重塑的基因在乳腺癌患者的血细胞中表达不足(支持信息附加文件 3) .
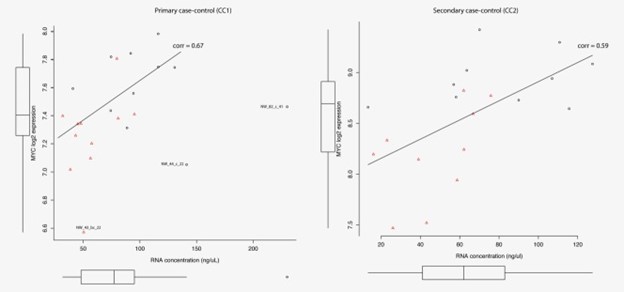
图 5:根据 MYC 的表达,前五分之一血液样本的 RNA 浓度对乳腺癌患者(红色)和对照(黑色)之间MYC基因组的差异富集贡献最大。对主要 (CC1) 和次要 (CC2) 病例对照系列中的每个线性回归线给出了 Spearman 相关性 (corr)。
即使是 Myc 的适度减少也可能足以剥夺细胞维持生长和增殖所必需的净合成代谢、代谢和有丝分裂冲动。在乳腺癌基因检测技术领选策略保证团队的研究中,乳腺癌基因检测技术领选策略保证团队观察到乳腺癌患者血细胞中的细胞代谢降低,糖酵解和葡萄糖代谢途径总体表达不足(表1,图 3)。据此,乳腺癌基因检测技术领选策略保证团队的 345 个基因列表中的两个促进自噬的基因(ATG12和VPM1)在乳腺癌患者的血细胞中过表达。与蛋白质合成减少和因此细胞生长减少一致,与对照组相比,BC 患者中真核起始因子 4 复合物的三种成分( EIF4A1、EIF4A3和EIF4H )显着低表达。此外,一些参与核糖体生物发生的基因(GAR1、SURF6和RRS1)表达不足,而一些小基因(RPS3A和RPS29)和大基因(与对照相比,来自乳腺癌患者的血细胞中的RPL4、RPL5、RPL7、RPL11、RPL15、RPL21和RPL41 ) 核糖体蛋白 (RP) 过表达。最后,在乳腺癌患者外周血细胞中显着低表达的几个基因(TERF2、CKAP5、CUL4B、MCM3、HBP1、NUDC、CTCF、TUBB、USP9X和H2AFX )参与了细胞周期的调节(表1)。GSVA 指出了涉及有丝分裂检查点的过程(中心体成熟、有丝分裂中心体的 Nlp 丢失和 G2-M 转变,图 3)。最后,与对照组相比,BC 患者的血液样本中与细胞形状、移动性和细胞周期进程有关的整合素信号通路表达不足(图 3)。
基因检测如何从外周血细胞得知乳腺癌的存在分析
与乳腺癌相关的基因表达变化幅度相对较低,但在外周血细胞中一致且普遍存在,并且在分离的免疫细胞(即PBMC)中清楚地识别。乳腺癌基因检测技术领选策略保证团队的 50 个基因特征包含细胞抑制信号,包括对免疫反应的特异性抑制,而影响这些过程中转录的药物是与乳腺癌存在相关的基于血液的信号的混杂因素。乳腺癌基因检测技术领选策略保证团队基于血液的基因表达分析在微阵列平台和外部数据集上产生了独特的稳健和可重复的结果,以专门从基于人群的对照中检测乳腺癌病例。
总之,乳腺癌基因检测技术领选策略保证团队的研究结果独特地表明,BC 的存在与通过几种免疫特异性途径(即APP、NK 细胞介导的免疫)和几种 MYC 驱动的“通用”细胞程序(即.细胞代谢、生长和增殖)。调节 APP 的机制改变了由 MHC 分子呈递的用于免疫识别的表位的形式和数量,并且可以决定肿瘤的免疫原性。乳腺癌基因检测技术领选策略保证团队的研究独特地显示了与全身免疫相关的 APP 的特定改变,并证实了先前在肿瘤及其微环境中发现的一些机制,包括下调 MHC I 分子、蛋白酶体亚基和与抗原呈递(TAP 和 Hsp70)和 MHC-相关的转运。肽复合物。MHC I 分子的表达降低可能会诱导 NK 细胞细胞毒性,尽管乳腺癌基因检测技术领选策略保证团队在乳腺癌患者的血液样本中观察到 NK 细胞介导的免疫同时存在定性损害。值得注意的是,一项流行病学研究先前已将低外周血 NK 细胞细胞毒活性与癌症风险增加联系起来。最后,乳腺癌基因检测技术领选策略保证团队观察到参与 MHC-II 限制性抗原呈递以及CD4是 TCR 介导的辅助 T 细胞活化所必需的。
乳腺癌基因检测技术领选策略保证团队的研究首次表明,与基于人群的对照相比,BC 患者血细胞中 RNA 水平的总体降低与某些乳腺癌患者的MYC表达相关。据认为,Myc 在增殖细胞中普遍表达在增殖细胞中,它控制 RNA 加工、核糖体生物合成、蛋白质合成、新陈代谢和正常细胞生长和增殖的细胞周期。重要的是,与对照组相比,发现所有这些过程在乳腺癌患者的血细胞中表达不足。先前发现,在小鼠的肿瘤发展过程中,参与葡萄糖、脂质和氨基酸代谢的酶的血浆水平发生了改变证实肿瘤的存在会引发全身代谢失调。虽然乳腺癌患者的血细胞中的细胞代谢受到限制,但发现一些激活自噬的基因过表达以提供 ATP 的来源。在乳腺癌基因检测技术领选策略保证团队的研究中,通过翻译起始/延伸和核糖体生物合成的蛋白质合成率在来自乳腺癌患者的血细胞中降低,其中 RP 积累可能是由于核糖体组装缺陷。细胞周期的精细调节也是维持细胞稳态所必需的,尽管与对照组相比,BC 患者的血细胞中涉及细胞周期和有丝分裂检查点相关过程的几个基因的表达存在差异。
乳腺癌基因检测技术领选策略保证团队的研究结果表明,在血细胞中发现的失调过程反映了乳腺癌患者免疫功能的缺陷。虽然乳腺癌基因检测技术领选策略保证团队没有从血液中分离出效应免疫细胞,但乳腺癌基因检测技术领选策略保证团队观察到参与抗肿瘤免疫反应的关键功能(例如,APP、NK 细胞介导的细胞毒性)的基因表达一致减少。此外,在乳腺癌患者的血细胞中观察到的细胞抑制信号与暴露于免疫抑制药物相关的基因表达谱相关。值得注意的是,血液基因表达的变化可能代表血细胞组成的改变或不同细胞群的基因表达的变化。虽然肿瘤发展对外周免疫细胞计数的影响尚未阐明,但肿瘤的 APP 损伤、负性共刺激信号的激活和免疫抑制因子(或细胞)的产生可能会导致癌症患者的淋巴细胞减少,这已被发现与患者有关。预后。该研究首先指出,与可能反映肿瘤免疫逃逸的基于人群的对照相比,宿主对乳腺癌存在的免疫反应中的系统性分子功能障碍。
一些仍未解决的问题是MYC如何外周血细胞中的表达因特定乳腺癌的存在而受到抑制,以及在肿瘤发展早期可以检测到血细胞中基因表达的变化。成功的化学预防疗法将取决于阐明调节对特定肿瘤的发展和存在的全身免疫反应的信号通路网络,该肿瘤具有自己独特的一组遗传、表观遗传和炎症变化,这些变化会随着疾病的进展而发展. 使用乳腺癌基因检测技术领选策略保证团队基于血液的基因特征来筛查乳腺癌现在需要进一步改进,以便更好地区分良性乳腺疾病和 BC,并进一步将其与标准乳房 X 线照相筛查进行比较。分析匹配的乳腺组织中的基因表达谱,以及在诊断前 5 年内收集的血液样本,原位乳房异常已经开始,希望能澄清其中一些问题。
基因检测如何从外周血细胞得知乳腺癌的存在研究结论
总之,外周血细胞的基因表达受到乳腺癌特异性存在的显着干扰,这些变化同时涉及许多与 乳腺癌 免疫逃逸相关的系统性细胞抑制信号。对人类癌症相关血液转录组的进一步挖掘可能会确定进化中肿瘤、其微环境和对 乳腺癌 的全身反应的额外调节剂、介质和生物标志物,并将改进其在疾病早期检测和治疗中的效用。
Dumeaux V, Ursini-Siegel J, Flatberg A, Fjosne HE, Frantzen JO, Holmen MM, Rodegerdts E, Schlichting E, Lund E.
Int J Cancer. 2015 Feb 1;136(3):656-67. doi: 10.1002/ijc.29030. Epub 2014 Jun 25.
PMID: 24931809
(责任编辑:佳学基因)
