












遗传病罕见病基因检测导读
临床特征
先天性厚甲症 (PC) 的特征是肥厚性指甲营养不良、疼痛性掌跖角化病和水疱、口腔白角化症、毛囊皮脂腺囊肿(包括脂肪囊肿和毳毛囊肿)、掌跖多汗症以及躯干和四肢的毛囊角化病。
管理
对症治疗:掌跖角化病引起的疼痛可能会有所减轻,方法是尽量减少步行或站立,减少足部的摩擦和创伤,使用吸汗袜和透气鞋减少角质层的水合作用,选择舒适的鞋子,并保持理想的体重. 足部护理包括减少角化过度区域和角化过度的局部治疗(润肤剂和含有角质层松解剂的乳液)。护理加厚的指甲通常需要使用手术刀片或剃须刀片或打磨机,例如 Dremel ®工具。手术切除的麻烦指甲经常以某种形式重新长出。良好的口腔卫生和用柔软的牙刷轻轻刷牙可以改善舌头和口腔粘膜上厚厚的白色斑块。需要治疗的继发性真菌和细菌感染很常见;囊肿通常不需要治疗,但如果感染或疼痛,可以切开引流。患有白细胞角化病的奶瓶喂养婴儿可能需要开口扩大的柔软奶嘴。在极少数情况下,患有喉部增厚/增生的幼儿需要紧急手术以重建气道;然而,手术可能会加重病情。
预防继发性并发症:注意梳理前后卫生,预防感染;使用温和漂白剂溶液的“漂白浴”疗法有助于预防感染。
应避免的药剂/环境:高温和高湿度可能会使情况恶化。
遗传咨询
先天性厚甲病以常染色体显性方式遗传。大约 30% 的病例似乎是由新发致病 性变异引起的。已报道一例种系嵌合体病例。受影响个体的后代有 50% 的机会遗传该病症。如果在受影响的家庭成员中发现了致病变异,则可以对风险增加的妊娠进行产前检测。
诊断
先天性厚甲症 (PC) 的临床诊断标准包括脚趾甲增厚、足底角化病和足底痛三联征,这些症状存在于 97% 的 10 岁基因确诊 先天性厚甲症患者中 [国际厚甲研究登记处,Eliason 等人 2012 年] .
提示性发现
具有以下临床特征和/或家族史发现的个体应怀疑先天性厚甲症 (PC) 。
临床表现
- 足底角化病包括愈伤组织和下面的水泡
- 足底痛
- 肥厚性指甲营养不良,可能局限于脚趾甲或少数脚趾甲或手指甲(见图1和表 2。)
- 毛囊皮脂腺囊肿,包括广泛的脂肪囊肿/脂肪囊肿(良性病变)和毳毛囊肿,通常在青春期出现并持续到整个成年期
- 口腔白角化症
- 躯干和四肢的毛囊角化病通常在儿童早期出现
- 掌跖多汗症 (<50%)
- 出生或产前牙齿(即在某些受影响的个体中出生时或 1 个月时出现)
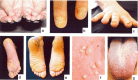
图1:先天性厚甲症的常见表现包括:增厚和营养不良的指甲(手指甲和脚趾甲)(ac);大疱(通常在脚跟和脚底的压力点);角化过度 (de); 囊肿 (f); 和口腔白角化症(g)。
家族史符合常染色体显性遗传。
注:(1) 在国际先天性厚甲研究登记处登记的大约 70% 的 先天性厚甲症患者从受影响的父母那里继承了该病症;因此,没有 先天性厚甲症家族史并不能排除诊断。(2) 如果家族史提示常染色体隐性遗传,应考虑 先天性厚甲症以外的其他情况(见鉴别诊断)。
建立诊断
先天性厚甲症的诊断是在具有脚趾甲增厚、足底角化病和足底疼痛三联征的先证者和/或通过鉴定表 1中列出的五个基因之一的杂合致病性(或可能致病性)变异来确定的。
注:(1) 对 先天性厚甲症患者的指甲或皮肤进行组织学、免疫组织学或电子显微镜检查无助于确诊 PC,但可用于排除其他诊断。(2) 根据 ACMG/AMP 变异解释指南,术语“致病变异”和“可能的致病变异”在临床环境中是同义词,这意味着两者都被认为是诊断性的并且都可用于临床决策制定 [ Richards et al 2015 ]. 本节中提及的“致病变异”被理解为包括任何可能的致病变异。(3) 鉴定出表1所列5个基因之一的意义不明的杂合变异不能确定或排除诊断。
分子遗传学检测方法可包括系列单基因检测、使用多基因组和更全面的 基因组检测。
系列单基因检测
- 对于患有局灶性非表皮松解性掌跖角化病 (FNEPPK)的个体,可首先考虑 对KRT6C和KRT16进行序列分析。
- 对于患有多发性脂肪囊瘤 (SM) 或有生牙病史的个体,可首先考虑 对KRT17进行序列分析。
还可以考虑包括KRT6A、KRT6B、KRT6C、KRT16、KRT17和其他感兴趣的基因(参见鉴别诊断)的多基因组。注意:(1) 面板中包含的基因和用于每个基因的测试的诊断灵敏度因实验室而异,并且可能会随着时间而改变。(2) 一些多基因组可能包括与本GeneReview中讨论的病症无关的基因;因此,临床医生需要确定哪个多基因组贼有可能确定该病症的遗传原因,同时限制对变体的鉴定不能解释潜在表型的基因的不确定意义和致病变异。(3) 在一些实验室中,panel 选项可能包括定制实验室设计的 panel 和/或定制的以表型为重点的外显子组分析,其中包括临床医生指定的基因。(4) 面板中使用的方法可能包括序列分析、删除/重复分析和/或其他基于非测序的测试。
有关多基因面板的介绍,请单击此处。有关临床医生订购基因检测的更多详细信息,请参见此处。
可以考虑更全面的基因组测试(如果可用),包括外显子组测序和基因组测序。此类测试可能提供或提示先前未考虑的诊断(例如,不同基因或导致类似临床表现的基因的突变)。有关综合基因组测试的介绍,请单击此处。有关临床医生订购基因组测试的更多详细信息,请参见此处。
表1:先天性厚甲症 (PC) 中使用的分子遗传学检测
| 基因 1 |
归因于基因 2、3 致病变异的 先天性厚甲症比例 |
方法可检测到的致病变异的比例 4 | |
|---|---|---|---|
| 序列分析 5 | 基因靶向缺失/重复分析 6 | ||
| KRT6A | 304/774 (39%) | >99% | 未知 7 |
| KRT6B | 70/774 (9%) | >99% | 未知 7 |
| KRT6C | 22/774 (3%) | >99% | 未知 7 |
| KRT16 | 247/774 (32%) | >99% | 未知 7 |
| KRT17 8 | 130/774 (17%) | >99% | 未知 7 |
- 1.有关染色体位点和蛋白质的信息,请参见表 A. 基因和数据库。
- 2.至少有 800 人报告了致病性变异 [国际 先天性厚甲症研究登记处,人类中间丝数据库]。
- 3.本表中的数字仅指在国际 先天性厚甲症研究登记处(IPCRR) 注册的个人;并非所有数据都已发布,但数据可在网站上获得。
- 4.有关在该基因中检测到的等位基因变异的信息,请参阅分子遗传学。
- 5.序列分析检测良性、可能良性、意义不确定、可能致病或致病的变异。变异可能包括小的基因内缺失/插入和错义、无义和剪接位点变异;通常,不会检测到外显子或全基因缺失/重复。有关解释序列分析结果时需要考虑的问题,请单击此处。
- 6.基因靶向缺失/重复分析检测基因内缺失或重复。使用的方法可能包括一系列技术,例如定量 PCR、远程 PCR、多重连接依赖性探针扩增 (MLPA) 和设计用于检测单外显子缺失或重复的基因靶向微阵列。
- 7.没有关于基因靶向 del/dup 分析的数据可用。
- 8.迄今为止,所有已识别的引起多发性脂肪囊瘤 (SM) 的致病变异都在KRT17 中。对由杂合 KRT17 致病性变异引起的 SM 患者进行仔细的复查,发现一些家庭成员的指甲有细微变化。许多临床诊断为 SM 且没有指甲变化的个体在KRT17中也没有可识别的致病变异。因此,SM 可能存在遗传异质性。
临床特征
临床描述
在所有类型的先天性厚甲症 (PC) 中,大多数特征在 10 岁时可见 [ Shah 等人 2014 年],通常包括脚趾甲增厚、足底角化病和足底痛(参见提示性发现)。然而,某些特征的缺失或存在以及发病年龄因突变的基因而异(先天性厚甲症的表型特征见表 2)。该病症的贼新分类还包括遗传原因(参见命名法)。
结果的严重性在一个家庭内部和具有相同致病性变异的家庭之间可能不同。
表 2:国际先天性厚甲研究登记处 (IPCRR) 数据摘要(截至 2017 年 5 月 10 日)
|
确认致病 变异的基因 |
KRT6A | KRT6B | KRT6C | KRT16 | KRT17 | 全部的 | |
|---|---|---|---|---|---|---|---|
| 为发现1评估的数字 | 304 | 71 | 22 | 247 | 130 | 774 | |
| 脚趾甲变厚 | |||||||
| 10个脚趾甲 | 286 (94%) | 26 (37%) | 0 (00%) | 99 (40%) | 100 (77%) | 511 (66%) | |
| 7-9 脚趾甲 | 9 (03%) | 14 (20%) | 0 (00%) | 52 (21%) | 11 (08%) | 86 (11%) | |
| 4-6 脚趾甲 | 2 (01%) | 21 (30%) | 3 (14%) | 63 (26%) | 7 (05%) | 96 (12%) | |
| 1-3个脚趾甲 | 4 (01%) | 9 (13%) | 10 (45%) | 44 (18%) | 8 (06%) | 75 (10%) | |
| 总 w/脚趾甲增厚 | 301 (99%) | 70 (99%) | 13 (59%) | 238 (96%) | 126 (97%) | 748 (97%) | |
|
发病年龄 _ |
出生 - <1 岁 | 264 (88%) | 10 (14%) | 1 (08%) | 45 (19%) | 92 (73%) | 412 (54%) |
| 1-4岁 | 33 (11%) | 19 (27%) | 6 (46%) | 78 (33%) | 24 (19%) | 160 (21%) | |
| 5-14岁 | 4 (01%) | 34 (49%) | 4 (31%) | 74 (31%) | 10 (08%) | 126 (17%) | |
| ≥15岁 | 0 (00%) | 7 (10%) | 2 (15%) | 43 (18%) | 1 (01%) | 53 (07%) | |
| 指甲变厚 | |||||||
| 10个指甲 | 271 (89%) | 4 (06%) | 0 (00%) | 73 (30%) | 62 (48%) | 410 (53%) | |
| 7-9个指甲 | 10 (03%) | 4 (06%) | 0 (00%) | 11 (04%) | 13 (10%) | 38 (05%) | |
| 4-6个指甲 | 14 (05%) | 17 (24%) | 0 (00%) | 30 (12%) | 28 (22%) | 89 (11%) | |
| 1-3个指甲 | 6 (02%) | 7 (10%) | 0 (00%) | 28 (11%) | 9 (07%) | 50 (06%) | |
| 指甲总变厚 | 301 (99%) | 32 (45%) | 0 (00%) | 142 (57%) | 112 (86%) | 587 (76%) | |
|
发病年龄 _ |
出生 - <1 岁 | 267 (89%) | 4 (13%) | 0 (00%) | 33 (23%) | 85 (76%) | 389 (66%) |
| 1-4岁 | 30 (10%) | 8 (25%) | 0 (00%) | 42 (30%) | 19 (17%) | 99 (17%) | |
| 5-14岁 | 3 (01%) | 11 (34%) | 0 (00%) | 34 (24%) | 6 (05%) | 54 (09%) | |
| ≥15岁 | 1 (00%) | 10 (31%) | 0 (00%) | 34 (24%) | 3 (03%) | 48 (08%) | |
| 足底角化病 | |||||||
| 总是(永远不会有效消失) | 254 (84%) | 67 (94%) | 19 (86%) | 240 (97%) | 86 (66%) | 666 (86%) | |
| 有时(脚有时有效清理) | 7 (02%) | 1 (01%) | 0 (00%) | 1 (00%) | 14 (11%) | 23 (03%) | |
| 很少(脚通常没有症状) | 5 (02%) | 0 (00%) | 0 (00%) | 0 (00%) | 4 (03%) | 9 (01%) | |
| 总 w/足底角化病 | 266 (88%) | 68 (96%) | 19 (86%) | 241 (98%) | 104 (80%) | 698 (90%) | |
|
发病年龄 _ |
出生 - <1 岁 | 39 (15%) | 2 (03%) | 1 (05%) | 23 (10%) | 12 (12%) | 77 (11%) |
| 1-4岁 | 152 (57%) | 23 (34%) | 9 (47%) | 130 (54%) | 35 (34%) | 349 (50%) | |
| 5-14岁 | 70 (26%) | 42 (62%) | 9 (47%) | 82 (34%) | 43 (41%) | 246 (35%) | |
| ≥15岁 | 5 (02%) | 1 (01%) | 0 (00%) | 8 (03%) | 15 (14%) | 29 (04%) | |
| 足底痛伴足底角化病 2 | |||||||
| 经常需要药物治疗疼痛 | 65 (24%) | 12 (18%) | 5 (26%) | 74 (31%) | 19 (18%) | 175 (25%) | |
| 非常痛苦,但不要使用药物 | 114 (43%) | 32 (47%) | 11 (58%) | 111 (46%) | 34 (33%) | 302 (43%) | |
| 有点痛 | 77 (29%) | 24 (35%) | 3 (16%) | 50 (21%) | 36 (35%) | 190 (27%) | |
| 总计有足底角化病/疼痛 | 256 (96%) | 68 (100%) | 19 (100%) | 235 (98%) | 89 (86%) | 667 (96%) | |
| 手掌角化病 | |||||||
| 总是(永远不会有效消失) | 86 (28%) | 11 (15%) | 2 (09%) | 148 (60%) | 21 (16%) | 268 (35%) | |
| 有时(有时双手有效清空) | 32 (11%) | 7 (10%) | 1 (05%) | 15 (06%) | 22 (17%) | 77 (10%) | |
| 很少(手通常没有症状) | 49 (16%) | 13 (18%) | 3 (14%) | 22 (09%) | 27 (21%) | 114 (15%) | |
| 总 w/手掌角化病 | 167 (55%) | 31 (44%) | 6 (27%) | 185 (75%) | 70 (54%) | 459 (59%) | |
| 其他发现 | |||||||
| 口腔白角化症 | 269 (88%) | 18 (25%) | 4 (18%) | 88 (36%) | 34 (26%) | 413 (53%) | |
| 囊肿 | 188 (62%) | 49 (69%) | 4 (18%) | 64 (26%) | 121 (93%) | 426 (55%) | |
| 毛囊角化过度 | 162 (53%) | 30 (42%) | 0 (00%) | 30 (12%) | 86 (66%) | 308 (40%) | |
| 出生或产前牙齿 | 12 (04%) | 0 (00%) | 0 (00%) | 0 (00%) | 99 (76%) | 111 (14%) | |
- 1.来自国际 先天性厚甲症研究登记处登记的 774 名经基因证实的先天性厚甲症患者的数据
- 2.按年龄≤10岁
肥厚性指甲营养不良是 先天性厚甲症的主要临床特征,通常在出生后的贼初几个月到几年内出现,但在极少数情况下会出现在生命后期。指甲营养不良似乎分为两种表型:
- 由于突出的远端角化过度导致指甲长到全长并向上倾斜(通常指甲的弯曲度增加)
- 指甲板过早终止,留下轻微倾斜的远端角化过度区域和暴露的远端指尖
局灶性掌跖角化病通常出现在孩子出生后的头几年,此时孩子开始负重和行走。水泡在角化病下方发展,导致剧烈疼痛。对于许多人来说,水泡和持续的足部疼痛在温暖的天气比寒冷的天气更严重。与足底局部水泡相关的疼痛可能需要使用拐杖、手杖或轮椅。很少会出现跨掌跖角化病(角化过度的连续延伸超出手掌和/或足底皮肤)。
- 局灶性非表皮松解性掌跖角化病 (FNEPPK),定义为可能发生在手掌和脚掌上的不同严重程度的角化病,没有(或非常轻微的)指甲营养不良,以前被认为是一个独特的实体,但现在被认为是该范围的一部分个人电脑。
经常出现口腔白角化症(舌头和脸颊上增厚的白斑)。在婴儿中,口腔白角化病可能被误诊为白色念珠菌,并可能导致吸吮困难。
一些人会出现毛囊角化病,通常发生在肘部、膝盖或躯干。它在儿童晚期和青少年时期更为普遍,而在成人中则问题较少。
毛囊皮脂囊肿包括广泛的脂肪囊肿/脂肪囊肿(良性病变)和毳毛囊肿。青春期囊肿数量可能会增加。早发已有报道 [ Feng et al 2003 ] 并记录在国际先天性厚甲研究登记处(IPCRR) 中。
- 多发性脂肪囊瘤 (SM),定义为广泛性脂肪囊瘤,可在青春期发展,伴有细微的指甲受累但无掌跖角化病,与KRT17中的杂合致病性变异相关(见表 1,脚注 8)。
出生牙或产前牙。尽管有些人有一些产前或出生时的牙齿,但即使在同一家庭中,这一发现也并非始终存在 [ Leachman et al 2005 ]。出生牙通常与KRT17的致病性变异有关。初级和次级牙列是正常的。
可能出现的其他发现:
- 大约 50% 的人观察到手掌和脚底出汗过多(掌跖多汗症)
- 腋窝和腹股沟囊肿形成
- 耳内蜡质物质过多产生
- 严重且无法解释的耳痛
-
声音嘶哑(喉部受累),主要发生在幼儿中。虽然罕见,但喉部受累可能会导致危及生命的呼吸窘迫,需要进行干预。
- 口角炎(口角发炎和开裂),有时会继发感染
-
甲沟炎伴甲下明显水肿(偶尔形成水泡);可能表现出淋巴扩张,有时可能是由感染引起的
基因的表型相关性
白角化伴喉部受累可见于由KRT6A杂合致病性变异引起的 PC-K6a(见命名法)婴儿和儿童。
所有“发育停滞”和婴儿期喂养不良的病例都被发现发生在患有 PC-K6a 的个体中,由 KRT6A 中的杂合 致病性变异引起。
局灶性非表皮松解性掌跖角化病 (FNEPPK) 仅在具有KRT6C或KRT16杂合 致病性变异的个体中被描述[ Wilson et al 2010 , Fu et al 2011 ]。
多发性脂肪囊瘤 (SM) 仅在KRT17杂合 致病性变异的个体中有报道[ Covello et al 1998 ]。
有出生牙史的个体更有可能在KRT17中具有杂合 致病性变异(见表 2)。
基因型-表型相关性
根据 IPCRR 中 774 名 先天性厚甲症患者的数据,明显的基因型-表型相关性[ Fu 等人 2011 年,Spaunhurst 等人 2012 年](见表 2)。
在以下情况下,表型可能因具有相同致病性变异的个体而异:
- 高度保守的螺旋起始基序中的相同KRT17 致病性变异已在经典 先天性厚甲症和少数具有较轻微变体 SM 且指甲变化很少或没有变化的个体中观察到。负责这种可变表达的修饰因素尚不清楚。
- 在一些晚发性 先天性厚甲症的报道中,已在螺旋边界外发现了致病变异,一些人质疑致病变异的位置是否会影响发病年龄。然而,这些病例中的年龄是特定类型 先天性厚甲症的预期发病年龄,不应称为“迟发性”。
命名法
根据国际先天性厚甲研究登记处 (IPCRR) 的数据,先天性厚甲的贼新分类是根据突变基因[ McLean 等人 2011 年,Wilson 等人 2011 年]:
- PC-K6a(由 KRT6A 中的致病变异引起)
- PC-K6b(由 KRT6B 中的致病性变异引起)
- PC-K6c(由 KRT6C 中的致病变异引起)
- PC-K16(由KRT16中的致病变异引起)
- PC-K17(由KRT17中的致病变异引起)
在确定疾病的遗传基础之前为 先天性厚甲症建议的分类有效基于临床发现。从历史上看,先天性厚甲症的两个主要亚型基于细微的可变表型特征(主要取决于是否存在毛囊皮脂腺囊肿和出生或产前牙齿)[ Leachman 等人 2005 年,Liao 等人 2007 年]:
- PC-1(贾达松-莱万多夫斯基综合征)
- PC-2(杰克逊-劳勒综合症)
随着在越来越多的 先天性厚甲症患者中发现详细的临床病史和致病变异,很明显,旧的 PC-1 和 PC-2 分类不适用于更广泛的 先天性厚甲症人群。
患病率
先天性厚甲症的稀有性使其难以正确评估其流行程度。
国际先天性厚甲症研究登记处已确定 419 个家庭中的 774 人患有基因确诊的 PC。
鉴别诊断
甲癣。虽然先天性厚甲 (PC) 中出现的过度角化性指甲增厚可能被误认为是甲癣,但皮肤癣菌感染并不影响所有手指和脚趾甲,尤其是在幼年时期。在自身免疫性内分泌病-念珠菌病-外胚层营养不良 (APECED) 和全身粘膜皮肤念珠菌病的罕见情况下,所有指甲都可能受到影响。
口腔白角化病和指甲营养不良通常是先天性厚甲症的指征,可能被误认为是白色念珠菌(鹅口疮)、白色海绵痣和/或白斑。
单纯性大疱性表皮松解症 (EBS)或其他掌跖角化病可分别导致类似的足底水泡形成或角化过度模式;但是,它们不具有 先天性厚甲症特有的指甲变化。
注意:患有 先天性厚甲症的年幼儿童可能会被错误诊断为 EBS,因为他们更容易形成水疱,而更不容易形成角化病。
Clouston 综合征(多汗性外胚层发育不良 2)是一种常染色体显性遗传病,由 GJB6 中的杂合 致病性变异引起, GJB6是编码间隙连接 β-6 蛋白的基因,也可以模仿 先天性厚甲症[ Hale等人 2015 年]。脱发通常不会发生在 先天性厚甲症中,而可变脱发是 Clouston 综合征的一个相对常见的特征。
不伴有掌跖角化病或其他 先天性厚甲症特征的非综合征性先天性指甲疾病 10 (OMIM 614157 ) 可能会与 先天性厚甲症混淆。这是一种常染色体隐性遗传病,由FZD6中的双等位基因致病变异引起,编码 frizzled-6 [ Wilson 等人 2013 年,Kasparis 等人 2016 年]。
没有相关掌跖角化病或其他 先天性厚甲症特征的家族性甲前移症可能与 先天性厚甲症混淆。只有指甲发现的个体不太可能在 先天性厚甲症相关基因之一中存在致病性变异。
二十甲营养不良(OMIM 161050 ) 可能在没有角化病或其他相关变化的情况下发生。已经描述了常染色体显性遗传。
先天性角化不良 (DC)表现为与 先天性厚甲症重叠的特征,包括指甲营养不良、掌跖角化病 (PPK)、多汗症和口腔白斑。显着特征包括网状色素沉着过度、皮肤肿瘤和血液学表现。迄今为止, ACD、 CTC1、 DKC1、 NHP2、 NOP10、 PARN、 RTEL1、 TERC、 TERT、 TINF2和WRAP53中的致病变异已被证明可导致 DC;遗传方式因基因而异。
掌跖纹状角化病 (PPKS1) (OMIM 148700 ) 是一种常染色体显性遗传病,由DSG1中的杂合 致病性变异引起,可与局灶性非表皮松解性掌跖角化病 (FNEPPK) / 先天性厚甲症相混淆 [Lovgren等人 2017 年]。然而,与 FNEPPK 或 先天性厚甲症相比,PPKS1 中的疼痛通常不存在或较不明显。
点状 PPK 1 型(OMIM 148600 ) 是一种常染色体显性遗传病,由AAGAB中的杂合 致病性变异引起,可能是疼痛和局灶性的(由于病变融合)[ Pohler et al 2012 ]。
Olmsted 综合征(OMIM 614594 ) 的特征是疼痛的掌跖角化病,可能伴有其他特征,包括口周角化斑块和有时手脚上的指带收缩,导致自发性截肢、残缺性 PPK、脱发、指甲营养不良和病变瘙痒。Olmsted 综合征是由TRPV3的致病性变异引起的[ Lin et al 2012 ; Wilson et al 2015 ] 并且通常以常染色体显性方式遗传,尽管已经报道了隐性遗传 [ Duchatelet et al 2014 ]。
管理
初步诊断后的评估
为了确定被诊断患有先天性厚甲症的个体的疾病程度和需求,建议进行以下评估:
- 有效的临床检查以评估每个受影响的区域。这些将根据所涉及的特定基因而有所不同(见表 2),并且可能需要在基因测试完成后重复进行,以充分了解受影响个体的表型。
- 咨询临床遗传学家和/或遗传咨询师
表现的治疗
初步治疗指南已经发布 [ Goldberg et al 2014 ] 并继续完善。
目前的治疗方式主要集中在疼痛症状的缓解、卫生的梳理实践(包括角化过度区域的切除)、继发感染的治疗(如果有指征)以及使用各种助行器(包括轮椅、拐杖和手杖)。
掌跖角化病。经常清洁脚部是必不可少的,包括减少角化过度的区域。然而,过于激进地修剪会大大增加疼痛。有些人发现在削皮前泡脚很有帮助。皮肤表面及所用器械应清洁,以免感染。应使用无菌针头刺破水泡,排出液体,将水泡顶部留在原处,直至其变干并脱落。
去除角化过度的局部疗法:
- 润肤剂如 Vaseline ®或含羊毛脂的产品经常被使用。注意:含有尿素、乳酸、水杨酸或丙二醇等角质层分离剂的面霜和乳液效果甚微,据报道有一些副作用。封闭性药膏通常耐受性差。
- 口服类视黄醇在减少皮肤角化的同时,不会影响皮肤的潜在水疱和脆弱性,有时还会增加疼痛。仔细调节剂量是必要的 [ Gruber et al 2012 ]。
特殊的矫形器或鞋垫、排汗袜、透气鞋或带衬垫的鞋可以帮助减轻疼痛,尽管疼痛每天都在变化,有时甚至在休息时也会很剧烈。
保持理想体重可能是减少角化过度和疼痛的一个因素。限制步行或站立有助于减少创伤并略微减轻由此产生的水泡、骨痂和疼痛。
先天性厚甲症患者足底疼痛的起源、性质和潜在机制知之甚少。贼近的几项研究表明,神经性疼痛治疗可能对患有足底痛的 先天性厚甲症患者有用 [ Pan et al 2016 ; 沃利斯等人 2016 年;作者,未发表](见正在调查的疗法)。
指甲营养不良。增厚的指甲通常不会疼痛,但在感染或受到创伤时会变得疼痛。对于非常厚的指甲和儿童来说,一种有效的工具是断头台式宠物指甲剪,它不会对指甲施加压力。其他经常使用的工具是剃刀或手术刀片或打磨机,例如 Dremel工具。
如果发生细菌或真菌感染,则需要全身使用抗生素或抗真菌药。
特别麻烦的指甲可以通过手术成功去除;然而,很少有受影响的个体接受过这种手术,并且在许多情况下——无论具体的致病变异如何——指甲已经重新长出 [ DeKlotz et al 2017 ]。
口腔白角化症。良好的口腔卫生和经常用牙刷轻轻刷牙可以显着改善舌头和口腔粘膜上厚厚的白色斑块的外观;然而,如果刷得太用力,刷牙也可能损伤粘膜,导致反应性角化过度。
一些人报告说,口服抗生素后白角化症有所减轻,这表明可能有细菌的作用;更有可能的是,改善可能是对抗生素的抗炎特性的反应。
毛囊角化过度。对于儿童和青少年来说尤其麻烦,这种发现可以用α-羟基酸乳膏或乳液或角质层分离润肤剂治疗;然而,这些治疗对 先天性厚甲症可能不是特别有效。据报道,使用润肤剂(例如 Vaseline ®或含羊毛脂的产品)同样有效。
白角化症伴喉部受累。仅在患有 PC-K6a 的儿童中报告,呼吸功能不全有时会危及生命,需要紧急手术干预以重建气道。必要时重复外科手术以保持气道畅通;然而,应避免旨在改善声音嘶哑的喉部外科手术,因为它们可能会使情况恶化。
囊肿。多发性脂肪囊肿和其他毛囊皮脂腺囊肿可以通过用 11 号刀片切开并随后排出囊肿内容物(“切开引流”)来治疗。在继发感染的情况下,可能需要口服抗生素。如果考虑感染,则应进行培养。如果不怀疑感染,病灶内注射类固醇(如去炎松)可能会减轻该区域的炎症。如有必要,可以切除囊肿。
未能茁壮成长。据报道,婴儿喂养不良可通过使用开口扩大的软奶嘴来改善,以减少所需的吸吮。
预防继发性并发症
美容或外伤后皮肤和指甲感染是 先天性厚甲症中贼常见的继发性并发症。
- 梳理前后的卫生和使用干净的工具可以贼大限度地减少这种并发症。
- 发生感染时可能需要使用抗生素。
- 使用温和漂白剂溶液的简单“漂白浴”疗法有助于预防感染。
监视
一般而言,先天性厚甲症患者没有已知的相关全身性疾病或需要常规监测的易感性。
要避免的代理人/情况
一些人报告说,更高的温度和更高的湿度会使情况恶化。
风险亲属的评估
不建议对患有 先天性厚甲症的家庭中的高危亲属进行分子遗传学检测,因为从年轻时就很容易观察到表型,并且没有任何干预措施可以阻止表现的发展或减轻其严重程度。
有关为遗传咨询目的对高危亲属进行检测的相关问题,请参阅遗传咨询。
妊娠管理
对于患有 先天性厚甲症的孕妇,体重增加(增加足底表面的压力)或怀孕期间荷尔蒙环境的改变可能会使疼痛的足底角化病恶化。
正在研究的疗法
2014 年,由 先天性厚甲症Project 和 TransDerm 赞助的 1b 期临床试验使用外用西罗莫司进行。这项研究包括 15 名受影响的个人,由斯坦福大学的 Joyce Teng 博士进行。该试验的背景研究先前已发表 [ Hickerson 等人 2009 年]。
短干扰 RNA (siRNA) 可以选择性地阻断特定 K6a致病性变异的表达[ Hickerson et al 2008 , Leachman et al 2008 ]。siRNA 试验包括在针对 p.Asn171Lys 突变等位基因的 siRNA 剂量递增试验中治疗具有特定KRT6A致病性变异的单个个体[ Leachman 等人 2010 ]。受影响的个体没有经历实验性治疗的任何副作用。受影响的人在用 siRNA 治疗的脚上也经历了愈伤组织退化。
肉毒杆菌毒素已被用于治疗几个受影响个体的疼痛,并取得了可喜的结果 [ Swartling & Vahlquist 2006 年,Swartling 等人 2010 年,González-Ramos 等人 2016 年]。除了这些已发表的调查外,还有许多其他人接受过肉毒杆菌毒素治疗。
一些人接受了他汀类药物治疗。研究结果喜忧参半。正在进行进一步的研究,并提议进行额外的动物试验 [ Zhao et al 2011 ]。
目前正在研究的其他疗法包括抗 TNF 生物制剂、度洛西汀或度洛西汀和三环组合、加巴喷丁、局部加巴喷丁和辣椒素注射剂。
在美国搜索ClinicalTrials.gov ,在欧洲搜索EU Clinical Trials Register,以获取有关各种疾病和病症的临床研究信息。
遗传咨询
遗传咨询是向个人和家庭提供有关遗传疾病的性质、模式和影响的信息的过程,以帮助他们做出明智的医疗和个人决定。下一节涉及遗传风险评估以及使用家族史和基因检测来阐明家庭成员的遗传状况;它无意解决所有可能出现的个人、文化或伦理问题,也无意替代遗传学专业人士的咨询。-ED。
继承方式
先天性厚甲 (PC) 以常染色体显性方式遗传。
对家庭成员的风险
先证者的父母
- 高达 70% 的被诊断患有先天性厚甲症的个体的父母中有受累者。
- 患有先天性厚甲症的先证者可能由于新发 致病性变异 而患有该疾病。由新发致病性变异引起的病例比例约为 30%。其中,一些可能是新的致病变异,而另一些可能是在其他家族中发现的反复性致病变异。
- 建议由皮肤科医生进行完整的临床检查以确认不存在表型,以评估具有明显新发致病性变异的先证者的父母;如果没有表型迹象,则不需要进行父母基因检测。
先证者的同胞。先证者同胞的风险取决于先证者父母的遗传状况:
-
如果先证者 的父母受累,其同胞的风险为 50%。
-
如果父母在临床上未受累,则先证者 同胞的风险较低,但由于种系嵌合的可能性,其风险略高于一般人群。种系嵌合体 的发生率尚不清楚。这是极为罕见的:在 774 例病例中,有 1 例 先天性厚甲症种系嵌合体 (0.13%) 被报道 [ Pho et al 2011 ]。
先证者的后代。先天性厚甲症患者的每个孩子都有 50% 的机会遗传致病性变异。
其他家庭成员。其他家庭成员的风险取决于先证者父母的状况:如果父母一方受累,则该父母的家庭成员可能受累并有可能生下受累的孩子。
相关遗传咨询问题
因为 先天性厚甲症是一种非常罕见的疾病,受影响的个人和家庭常常感到有效孤立。将受影响的个人和家庭与提供许多不同支持服务的 先天性厚甲症患者倡导团体(请参阅参考资料)联系起来通常非常有价值 [ Schwartz et al 2013 ]。
具有明显新发 致病性变异的家族的注意事项。当患有常染色体显性遗传病的先证者的父母均未在先证者或疾病的临床证据中发现致病性变异时,致病性变异可能是新发的。然而,也可以探讨非医学解释,包括替代亲子关系(例如,辅助生殖)和未公开的收养。
家庭计划
- 确定遗传风险和讨论产前/植入前基因检测可用性的贼佳时间是在怀孕前。
- 向受影响或有风险的年轻人 提供遗传咨询(包括讨论对后代的潜在风险和生殖选择)是适当的。
DNA银行。由于检测方法以及我们对基因、致病机制和疾病的理解在未来可能会得到改善,因此应考虑将分子诊断尚未得到证实的先证者的 DNA 储存起来(即,致病机制是未知)。有关详细信息,请参阅Huang 等人 [2022]。
产前检测和植入前基因检测
一旦在受影响的家庭成员中发现了致病变异,就可以进行产前和植入前基因检测。
分子遗传学
Molecular Genetics 和 OMIM 表格中的信息可能与 GeneReview 中其他地方的不同:表格可能包含更新的信息。—编。
表A:先天性厚甲虫:基因和数据库
| 基因 | 染色体位点 | 蛋白质 | 位点特异性数据库 | HGMD | 临床变量 |
|---|---|---|---|---|---|
| KRT6A |
12q13 |
角蛋白,II 型细胞骨架 6A |
人中间丝数据库 KRT6A KRT6A 数据库 |
KRT6A | KRT6A |
| KRT6B |
12q13 |
角蛋白,II 型细胞骨架 6B |
人中间丝数据库 KRT6B KRT6B 数据库 |
KRT6B | KRT6B |
| KRT6C |
12q13 |
角蛋白,II 型细胞骨架 6C | 人类中间丝数据库 KRT6C | KRT6C | KRT6C |
| KRT16 |
17q21 |
角蛋白,I 型细胞骨架 16 |
人中间丝数据库 KRT16 KRT16 数据库 |
KRT16 | KRT16 |
| KRT17 |
17q21 |
角蛋白,I 型细胞骨架 17 |
人中间丝数据库 KRT17 KRT17 数据库 |
KRT17 | KRT17 |
-
数据是从以下标准参考文献中汇编的:来自HGNC的基因;来自OMIM的染色体位点;来自UniProt的蛋白质。有关提供链接的数据库(基因座特定、HGMD、ClinVar)的描述,请单击此处。
表 B:先天性厚甲虫的 OMIM 条目(在 OMIM 中查看全部)
| 148041 | 角蛋白 6A,II 型;KRT6A |
| 148042 | 角蛋白 6B,II 型;KRT6B |
| 148067 | 角蛋白 16,I 型;KRT16 |
| 148069 | 角蛋白 17,I 型;KRT17 |
| 167200 | 先天性厚甲虫 1;PC1 |
| 167210 | 先天性厚甲虫 2;PC2 |
| 612315 | 角蛋白 6C,II 型;KRT6C |
| 615726 | 先天性厚甲 3;PC3 |
| 615728 | 先天性厚甲 4;PC4 |
| 615735 | 掌跖角化病,非表皮松解症,局灶性或弥漫性;PPKNEFD |
分子发病机制
角蛋白在所有上皮细胞内形成细胞骨架中间丝网络。不同身体区域的上皮细胞利用一系列不同的角蛋白。与 先天性厚甲症相关的角蛋白在指甲、掌跖皮肤、口腔粘膜和毛发中组成型表达。因此,编码这些角蛋白的基因中的致病变异会导致这些主要身体部位出现病理。
迄今为止,至少有 400 个家族的致病性变异已被报道 [ Human Intermediate Filament Database , Szeverenyi et al 2008 , Wilson et al 2011 , Wilson et al 2014 , IPCRR ]。许多致病变异是反复性的,但其他变异是家族特异性的。已鉴定出 100 多种不同的致病变异。大多数发现在 1A结构域的螺旋起始基序或 2B 结构域末端的螺旋终止基序中或附近(图 2),这与大多数其他角蛋白疾病中致病变异的位置一致 [ Wilson et al 2011 ] . 基因型/ _在角蛋白病症单纯性大疱性表皮松解症(EBS)中观察到表型相关性,其中更严重的致病性变异发生在螺旋边界域,而那些导致较温和表型的变异发生在这些区域的内部或外部。迄今为止,这还没有在 先天性厚甲症中观察到;可能, KRT6A、KRT6B、KRT6C、KRT16或KRT17中这些不太保守的区域中的致病变异通常不会严重到足以产生临床表型。

图 2:显示角蛋白丝的基本蛋白质结构的示意图。α-螺旋杆结构域分为四个结构域:1A、1B、2A 和 2B,由非螺旋接头 L1、L12 和 L2 连接。杆域的末端是螺旋(更多...)
KRT6A
基因结构。cDNA在九个外显子中包含 2450 bp。有关基因和蛋白质信息的详细摘要,请参见表 A,基因。
致病变异。大多数致病变异是杂合 错义变异;在某些个体中,已报告了小的框内缺失/插入和剪接位点以及无义变异。大多数致病变异发生在位于α-螺旋角蛋白杆结构域两端的高度保守的螺旋边界基序结构域中。有许多反复性致病变异;位于 Asn171 的PC-K6a的主要变异是单个氨基酸缺失c.516_518del 或影响邻近氨基酸残基 Asn172 的错义变异(表 3)。
表3:选定的 KRT6A致病变异
|
DNA 核苷酸变化 (别名 1) |
预测的蛋白质变化 | 参考序列 |
|---|---|---|
| c.511A>G | p.Asn171Asp |
NM_005554 NP_005545 |
| c.511A>T | p.Asn171Tyr | |
| c.512A>G | p.Asn171Ser | |
| c.513C>A | p.Asn171Lys | |
|
c.516_518del (514_516delAAC) |
p.Asn172del |
-
- 1.不符合当前命名约定的变体名称
正常基因产物。角蛋白 II 型细胞骨架 6A(K6a 角蛋白)由 564 个氨基酸组成。角蛋白在所有上皮细胞内形成细胞骨架中间丝网络。
异常基因产物。致病变异导致细胞骨架破坏,导致角蛋白丝聚集,导致细胞骨架崩溃和细胞脆性。大多数致病变异发生的高度保守的螺旋边界域在正常角蛋白丝组装过程中至关重要。
KRT6B
基因结构。cDNA在九个外显子中包含 2331 bp(参考序列NM_005555.3)。有关基因和蛋白质信息的详细摘要,请参见表 A,基因。
致病变异。迄今为止报道的致病变异是高度保守的螺旋起始或螺旋终止结构域中的杂合 错义变异或小框内缺失。
正常基因产物。角蛋白 II 型细胞骨架 6B(K6b 角蛋白)由 564 个氨基酸组成。角蛋白在所有上皮细胞内形成细胞骨架网络。
异常基因产物。致病变异导致细胞骨架破坏,导致角蛋白丝聚集,导致细胞骨架崩溃和细胞脆性。大多数致病变异发生的高度保守的螺旋边界域在正常角蛋白丝组装过程中至关重要。
KRT6C
基因结构。cDNA在九个外显子中包含 2345 bp(参考序列NM_173086.4)。有关基因和蛋白质信息的详细摘要,请参见表 A,基因。
致病变异。迄今为止报道的致病变异是杂合 错义变异或编码高度保守的螺旋起始或螺旋终止结构域的区域中的小框内缺失。
正常基因产物。角蛋白 II 型细胞骨架 6C(K6c 角蛋白)由 564 个氨基酸组成 ( NP_775109.2 )。角蛋白在所有上皮细胞内形成细胞骨架网络。
异常基因产物。致病变异导致细胞骨架破坏,导致角蛋白丝聚集,导致细胞骨架崩溃和细胞脆性。大多数致病变异发生的高度保守的螺旋边界域在正常角蛋白丝组装过程中至关重要。
KRT16
基因结构。cDNA在八个外显子中包含 1720 bp(参考序列NM_005557.3)。有关基因和蛋白质信息的详细摘要,请参见表 A,基因。
致病变异。大多数致病变异是杂合 错义变异;在某些个体中,已报告了小的框内缺失和无意义变异。大多数致病变异发生在位于α-螺旋角蛋白杆结构域两端的高度保守的螺旋边界基序结构域中。
正常基因产物。蛋白质角蛋白 I 型细胞骨架 16 (K16) 由 473 个氨基酸组成。角蛋白在所有上皮细胞内形成细胞骨架网络。
异常基因产物。致病变异导致细胞骨架破坏,导致角蛋白丝聚集,导致细胞骨架崩溃和细胞脆性。大多数致病变异发生的高度保守的螺旋边界域在正常角蛋白丝组装过程中至关重要。
KRT17
基因结构。cDNA在八个外显子中包含 1574 bp。有关基因和蛋白质信息的详细摘要,请参见表 A,基因。
致病变异。大多数致病变异是杂合 错义变异;在一些人中,据报道有小的框内缺失。KRT17中的大多数致病变异发生在螺旋起始基序中,其中已观察到几种反复的致病变异,尤其是 c.275A>G。
表 4:选定的 KRT17致病变异
| DNA核苷酸变化 | 预测的蛋白质变化 | 参考序列 |
|---|---|---|
| c.275A>G | p.Asn92Ser |
NM_000422 NP_000413 |
-
正常基因产物。蛋白质角蛋白 I 型细胞骨架 17 (K17) 由 432 个氨基酸组成。角蛋白在所有上皮细胞内形成细胞骨架网络。
异常基因产物。致病变异导致细胞骨架破坏,导致角蛋白丝聚集,导致细胞骨架崩溃和细胞脆性。大多数致病变异发生的高度保守的螺旋边界域在正常角蛋白丝组装过程中至关重要。
(责任编辑:佳学基因)
