












【佳学基因检测】前额高腭弓高囟门增大人中长眼距过宽肌张力减退还有癫痫是不是遗传性DBP缺乏症?
罕见病、遗传病基因检测导读:
D-双功能蛋白 (DBP) 缺乏症是一种常染色体隐性遗传的过氧化物酶体脂肪酸氧化障碍。《人体生理功能及疾病表现的基因序列根源》指出双功能是指人体内的单一的蛋白质如酶包含多个活性结构域,负责过氧化物酶体 β-氧化的连续步骤。对于D-双功能蛋白(DBP)来说,DBP 催化超长链脂肪酸 (VLCFA) C26:0、支链脂肪酸(鹦鹉螺酸)和胆汁酸中间体的 β-氧化的第二步(水合)和第三步(脱氢) (二羟基胆甾烷酸 (DHCA) 和三羟基胆甾烷酸 (THCA))。DBP 包含三个结构域,由 4 型 17-β 羟基类固醇脱氢酶 ( HSD17B4)编码基因借助体内结构与功能蛋白的产生机制生成这一蛋白质。N 端短链乙醇脱氢酶结构域由外显子 1-12 编码,中心 2-烯酰辅酶 A 水合酶结构域由外显子 12-21 编码,C 端固醇载体蛋白 2 样结构域(SCP-2L ) 由外显子 21–24 编码。DBP 是一种具有 79 kD 亚基的同型二聚体酶。进入过氧化物酶体后,蛋白质被切割,产生一个 35 kD 的脱氢酶单元和一个 45 kD 的水合酶加 SCP-2L 单元。
DBP 缺乏症根据缺乏的活性分为三种亚型。DBP 缺乏症 I 型是 2-烯醇-CoA 水合酶和 3-羟酰基-CoA 脱氢酶活性的缺乏,DBP 缺乏症 II 型是单独的水合酶活性缺乏,DBP 缺乏症 III 型是单独的脱氢酶活性缺乏。佳学基因检测对超过 100 名 DBP 缺乏患者的临床和生化记录进分分析,总结了所有三种生化亚型患者的临床特征的相似性。几乎所有患者都在出生后的第一个月内出现肌张力减退和癫痫发作,超过三分之二的患者还表现出 Zellweger 样面部特征(即前额高、腭弓高、囟门增大、人中长、眼距过宽)。大多数患有 DBP 缺乏的婴儿 (>80%) 在 2 岁之前死亡,通常死于呼吸系统并发症。生化检测通常会发现血浆 C26:0、DHCA、THCA 以及鹦鹉螺酸及其前体植烷酸水平升高。只有少数 (<2%) 的 DBP 缺陷患者会显示正常的生化测试。因此,佳学基因强调在临床上怀疑患者是过氧化物酶体功能紊乱的情况下需要采用培养的皮肤成纤维细胞进行进一步基因解码研究的重要性。
佳学基因检测记录了两兄弟经过致病基因鉴定基因解码确诊患有 DBP 缺乏症的临床案例。虽然这些男孩表现出过氧化物酶体功能障碍的一些典型临床特征(听力障碍、小脑和感觉性共济失调),但临床检查没有出现血浆 VLCFA、鹦鹉螺酸和植烷酸或尿胆汁酸的明显异常。基于外显子组测序的基因检测对于检测 DBP 水合酶和脱氢酶结构域中的复合杂合突变至关重要。针对 DBP 活性的进一步成纤维细胞酶测试证实水合酶和脱氢酶活性显着降低但可检测到。据此,佳学基因致病基因鉴定基因解码认为患者有一种新型的 DPB 缺陷。学术界根据其独特的临床、生化和遗传特征将其定义为 IV 型,从而扩大了与 DBP 功能改变相关的临床表型谱。
患者病情介绍
患者 1
一名 16.5 岁的男孩在 3.5 岁时被诊断出语言表达迟缓和发音困难,这是由中度至重度感音神经性听力障碍引起的。他的语言发展在助听器的帮助下完全正常化。他早期的粗大和精细运动发育正常;他小时候会滑冰和打曲棍球。他在普通课堂环境中的学业成绩很好。11 岁时,他患上了隐匿的进行性步态共济失调。他被发现有双侧高弓足和轻度槌状趾足畸形、弥漫性反射消失和屈肌足底反应。注意到前间室肌肉非常轻度无力,因此使用了踝足矫形器。小纤维感官测试在库存分布中略有下降。振动觉和本体感觉保持完好。有明显的轻度不协调;快速手指运动缓慢,脚跟到胫骨测试受损。Romberg征阴性。神经传导研究(表1) 显示具有脱髓鞘特征的轻度感觉运动性多发性神经病(运动传导速度;20–25 m/sec;正常上肢≥50 m/sec,下肢≥40 m/sec)。感觉和运动神经振幅正常,但腓总神经 CMAP 振幅较低。随着时间的推移,他的高弓足恶化,他的神经传导研究显示进行性脱髓鞘(潜伏期延长增加)和长度依赖性轴突丧失(运动和感觉振幅进行性丧失)的证据。15.5 岁时的眼底镜检查显示广泛的周边视网膜萎缩伴中央黄斑(即严重的视杆细胞功能障碍伴视锥细胞相对保留)与色素性视网膜炎一致(图 1)。视力正常。视觉诱发电位正常。暗视杆状视网膜电图 (ERG) 显示轻度异常,表明周边视网膜的杆状细胞功能障碍。明视锥和 30 Hz 闪烁也异常,表明视锥功能障碍。在他 16.5 岁时的最后一次临床随访中,他在家中可以进行短距离走动,但由于他严重的共济失调,长距离走动需要轮椅。他没有表现出生长或青春期延迟的迹象。他的身高在 10-25百分位之间,并且在 16 岁时,他被注意到具有与年龄相适应的青春期发育;Tanner IV 阴毛和 12-15 mL 睾丸体积。他的认知、语言和视觉都完好无损。他没有肾脏或肝脏受累的临床或生化证据。
表1:神经传导基因解码
|
|
正常 |
患者 1 |
患者 2 |
||
|---|---|---|---|---|---|
|
|
|
就诊年龄 |
|
||
| 12年 | 13年 | 16年 | 13 ½ 年 | ||
|
运动: |
|
|
|
|
|
|
正中神经 |
|
|
|
|
|
|
DML(手腕到 APB) |
< 4.2 |
|
3.6 |
5.4 |
3.4 |
|
CMAP (毫伏) |
≥3.9 |
|
9.2 |
4.1 |
8.6 |
|
CV(米/秒) |
> 47 |
|
27 |
27 |
33 |
|
尺神经 |
|
|
|
|
|
|
DML(毫秒;手腕到 ADM) |
< 3.4 |
2.6 |
3.1 |
3.6 |
2.2 |
|
CMAP (毫伏) |
≥5.9 |
13.1 |
12.7 |
4.6 |
7.7 |
|
CV(米/秒) |
> 47 |
25 |
27 |
23 |
24 |
|
胫神经 |
|
|
|
|
|
|
DML(毫秒;脚踝到 AH) |
< 6.0 |
5.3 |
6.2 |
10.5 |
4.5 |
|
CMAP (毫伏) |
≥3.9 |
3.3 |
3.6 |
0.5 |
6.6 |
|
CV(米/秒) |
> 39 |
21 |
22 |
15 |
26 |
|
腓神经 |
|
|
|
|
|
|
DML(毫秒;脚踝到 EDB) |
< 6.0 |
5.0 |
|
8.5 |
4.5 |
|
CMAP (毫伏) |
≥ 2.4 |
1.7 |
|
0.8 |
2.9 |
|
CV(米/秒) |
> 39 |
22 |
|
17 |
43 |
|
感官: |
|
|
|
|
|
|
正中神经 |
|
|
|
|
|
|
PL(毫秒;手腕到手指-II) |
< 3.2 |
2.5 |
2.8 |
3.4 |
2.1 |
|
SNAP (μV) |
≥ 14 |
67 |
69 |
15 |
36 |
|
CV(米/秒) |
|
46 |
39 |
40 |
52 |
|
尺神经 |
|
|
|
|
|
|
PL(毫秒;手腕到手指-V) |
< 3.3 |
2.0 |
2.2 |
3.1 |
2.2 |
|
SNAP (μV) |
≥ 9 |
43 |
64 |
26 |
44 |
|
CV(米/秒) |
|
47 |
50 |
35 |
43 |
|
腓肠神经 |
|
|
|
|
|
|
PL(毫秒;小腿到拉特购物中心) |
< 4.2 |
3.8 |
3.0 |
NR |
2.7 |
|
SNAP (μV) |
≥ 5 |
11 |
17 |
NR |
9.5 |
| CV(米/秒) | 42 | 40 | NR | 35 | |
图例:DML=远端开始运动潜伏期;CMAP=复合运动动作电位;CV=传导速度;PL=峰值起始潜伏期;APB=外展拇短肌;ADM=小指外展肌;AH=拇外展肌;EDB=趾短伸肌;NR=无响应。
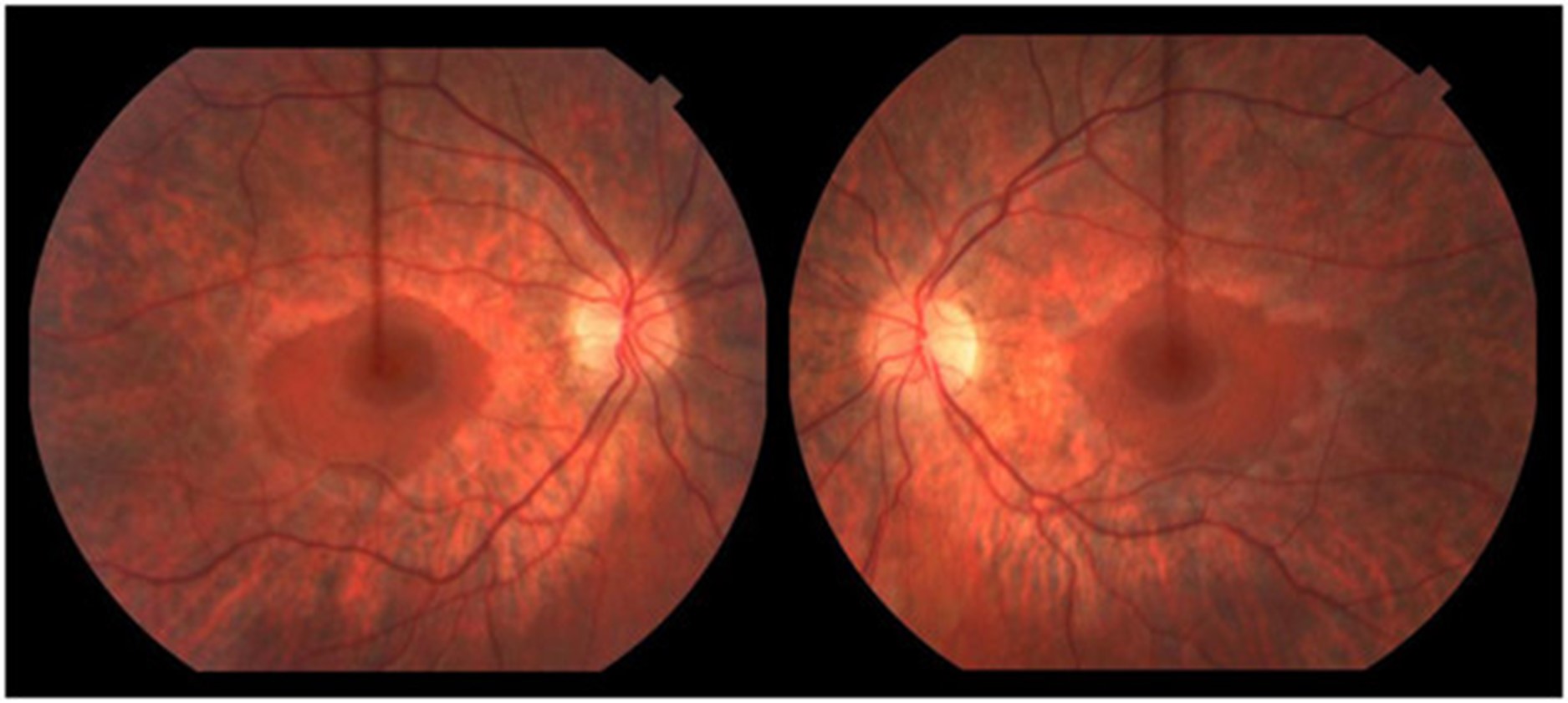
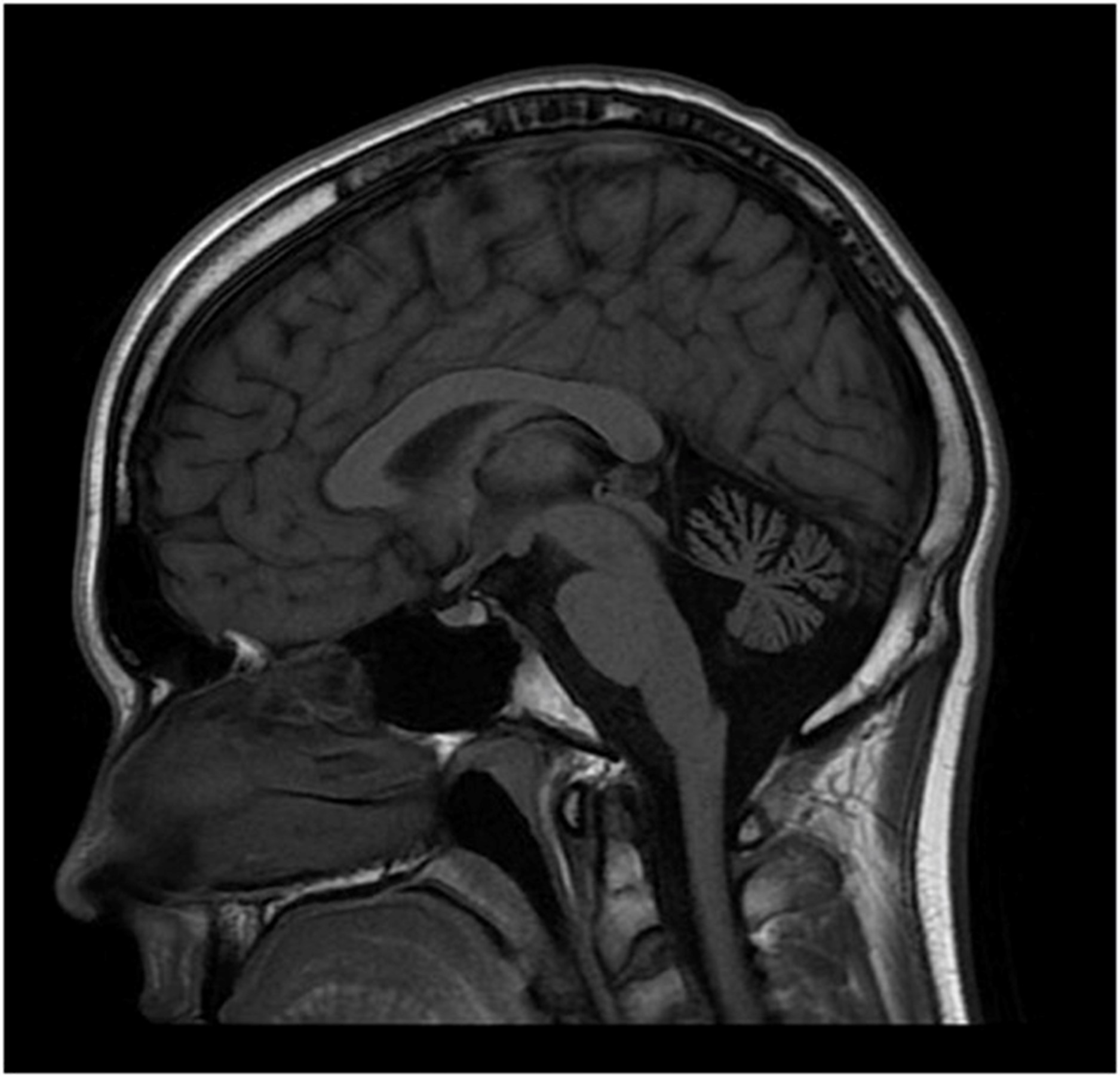
12 岁时脑部 MRI 显示小脑萎缩(图2). 在几年的时间里进行了广泛的基因检测和代谢测试,包括正常PMP22重复/缺失分析基因检测、MPZ、GJB1、PMP22和c10orf2基因检测、脊髓小脑性共济失调组(SCA 1、2、3、6、7、8、17 ), Friedreich 共济失调重复基因检测和血清维生素 E 水平。染色体微阵列基因检测正常。肌肉活检(15 岁时)显示轻微的神经源性变化。肌肉呼吸链酶检测正常。血浆肾上腺和睾丸性类固醇水平正常。
在家族史分析时,患者1的弟弟具有非常相似的特征。患者的父母、姐姐都没有这种疾病。对血缘关系进行扩展调查,没有发现其他患者。佳学基因亲缘亲系检测证明,患者父母不是近亲,没有血缘关系。
病人 2
这位 14 岁的弟弟在 2 岁时被确诊患有中度至重度感音神经性听力损失,需要助听器。他的早期神经发育正常。他仍然很活跃;最后随访能够滑冰 3 公里和越野滑雪 1-1.5 小时。他不需要踝足矫形器或任何其他辅助设备。他的检查对非常轻度的高弓足和反射减退(二头肌 1+、肱桡肌 1+、三头肌 2+、髌骨 1+、踝反射 0)、屈肌底反应和轻度前间室无力(胫骨前肌 4+/5、腓骨肌)具有重要意义长肌 4+,拇长伸肌 4)。除了脚趾针刺感觉过敏外,感官测试正常。注意到轻微的脚踝紧绷。协调是正常的。神经传导研究(表1) 显示具有脱髓鞘特征的轻度感觉运动性多发性神经病。眼底检查显示广泛的周边视网膜萎缩,中央黄斑不受累。视力和 ERG 正常。他的认知完好无损,并且在常规课堂环境中取得了良好的学业成绩。尽管他的身高一直低于第 5个百分点,但他表现出正常的生长速度。在将近 14 岁的时候,他没有显示出阴毛初现的迹象,Tanner I 阴毛和 5 mL 睾丸体积。他的实际年龄为 13 岁 8 个月,骨龄为 11 岁,符合体质性生长和青春期延迟。他没有显示肾脏或肝脏受累的证据。
致病基因鉴定基因解码基因检测
佳学基因对两兄弟的 DNA 进行了外显子组捕获和高通量测序。按照标准程序从血液中提取总基因组 DNA。使用 Agilent SureSelect 50Mb 全外显子试剂盒进行外显子组靶点富集,测序 (Illumina HiSeq) 为每个样本生成 65 Gbp 的 100 bp 配对末端读数。编码序列区域 (CCDS) 的平均覆盖率,在考虑重复读取后,两个人分别为 237 倍和 212 倍。患者 1 中 91.5% 的 CCDS 碱基和患者 2 中 90.0% 的 CCDS 碱基覆盖率≥20 倍,两个人中 96.8% 的 CCDS 碱基覆盖率≥5 倍。内部注释管道用于调用和注释编码和剪接位点变体。使用 Picard 标记并排除重复读取。使用 SAMtools pileup和 varFilter调用单核苷酸变异和短插入和缺失 (indel),并进行质量过滤以要求至少 20% 的读数支持变异调用。使用 Annovar以及自定义脚本对基因突变序列进行注释,以选择编码和剪接位点变体,并排除 NHLBI 外显子组服务器中代表的常见(≥1% 次要等位基因频率)多态性,与佳学基因所记录的1000个对照外显子组序列进行比对。根据在两名受影响患者中发现的突变序列进行优先排序。鉴于假定的常染色体隐性遗传模式,仅考虑具有纯合或多个杂合突变的基因。
突变序列的基因检测验证
Sanger 测序用于验证下一代测序鉴定的突变,并评估家族中变异的分离。采集了血液样本,并从患病的兄弟和未受影响的父母和姐妹身上提取了 DNA。使用引物5'-GAGTGGATAGGTTGAGAATGTCAGTG-3 '和5'-TTTAGACAGACAGCCTTAGTCGGG-3 '进行 PCR以测试 c.101C>T 变体和5'- ACCAATAACCAGCCATGTTTCCT -3 '和5' - TCCTACCTTTCCATACCTTTGCAT - 3 '以测试c.101C>T 变体.1547T>C 变体。
生化分析
成纤维细胞中的生化测试如所述进行(表2) 。| 测试 | 单位 | 患者 1 | 病人 2 | 参考范围 |
|---|---|---|---|---|
|
等离子体: |
|
|
|
|
|
VLCFA浓度 |
|
|
|
|
|
C26:0 六烷酸 |
微克/毫升 |
0.220 |
0.270 |
0.23 ± 0.09* |
|
C26/C22 |
|
0.021 |
0.012 |
0.01 ± 0.004* |
|
C24/C22 |
|
0.918 |
0.967 |
0.84 ± 0.918* |
|
植烷酸 |
微克/毫升 |
1.260 |
0.810 |
< 3.0 |
|
紫杉酸 |
微克/毫升 |
0.140 |
0.060 |
< 0.3 |
|
成纤维细胞: |
|
|
|
|
|
过氧化氢酶免疫荧光 |
|
接近正常 |
普通的 |
普通的 |
|
VLCFA浓度: |
|
|
|
|
|
C26:0浓度 |
μmol/g蛋白质 |
0.20 |
0.20 |
0.18 - 0.38 |
|
C24:0浓度 |
μmol/g蛋白质 |
6.52 |
6.79 |
7.76 - 17.66 |
|
C22:0浓度 |
μmol/g蛋白质 |
3.00 |
3.23 |
3.84 - 10.20 |
|
比例 C26:0 / C 22:0 |
|
0.07 |
0.06 |
0.03 - 0.07 |
|
比例 C24:0 / C 22:0 |
|
2.18 |
2.10 |
1.55 - 2.30 |
|
过氧化物酶体功能: |
|
|
|
|
|
β-氧化(C16:0) |
pmol / (mg 蛋白质 / 小时) |
3876 |
5061 |
3330 - 7790 |
|
β-氧化(C26:0) |
pmol / (mg 蛋白质 / 小时) |
1290 |
1325 |
800 - 2040 |
|
β-氧化(鹦鹉螺酸) |
pmol / (mg 蛋白质 / 小时) |
157 |
248 |
790 - 1690 |
|
α-氧化(植烷酸) |
pmol / (mg 蛋白质 / 小时) |
38 |
45 |
28 - 95 |
|
D-双功能蛋白活性: |
|
|
|
|
|
水合酶 |
pmol / (mg 蛋白质 / 分钟) |
43 |
53 |
115 - 600 |
|
脱氢酶 |
pmol / (mg 蛋白质 / 分钟) |
2个 |
3个 |
25 - 300 |
|
免疫印迹: |
|
|
|
|
|
舒张压 79 kDa |
|
痕迹 |
痕迹 |
当下 |
|
舒张压 45 kDa |
|
缺席的 |
缺席的 |
当下 |
| 舒张压 35 kDa | ±/− | ±/− | 当下 |
粗体和下划线的值是异常的;*表示平均值 +/- 1 个标准差。
基因检测中的全外显子测序的突变位点验证结果
外显子组测序和变体验证
在HSD17B4(表3). c.1547T>C 被发现在一个覆盖率非常低的区域,在患者 2 的 8 个读数中仅出现 2 个(变异质量为 18.4%),因此只有在他兄弟的两个变异被注释后才能通过目视检查来识别. Sanger 测序证实两兄弟中存在 c.1547T>C 和 c.101C>T突变。父亲和母亲各含有中一个突变的杂合子;c.1547T>C; p.Ile516Thr来自母亲c.101C>T;p.Ala34Val来自父亲(图3). SIFT 算法预测这两种突变都是有害的(SIFT 得分分别为 0.03 和 0),并且采用 PolyPhen2预测也是破坏性的(HumDiv 得分分别为 0.999 和 0.997)。这两种突变分别被判断为致病性的;但是,以前从未见过它们的组合。
表3:过滤外显子组测序的基因突变
| 具有错义、无义、插入缺失或剪接变异的基因 | 6453 |
|---|---|
|
具有罕见突变的基因1 |
372 |
|
兄弟共有突变的基因 |
109 |
| 具有纯合子/多个杂合子突变的基因 | 2 2 |
1如果变异在 1000 个基因组数据库、NHLBI 外显子组服务器中具有 >0.01 的次要等位基因频率 (MAF),或者在我们中心测序的 435 个对照外显子组中 >2 个出现,则变异被过滤掉。
2 2个基因分别为C16orf82和FAM83C;这些基因被拒绝作为候选基因,因为两个对照样本在 C16orf82 中具有相同的序列变体,而一个对照样本在FAM83C中具有相同的序列变体。然后重新检查兄弟姐妹共有的至少一个突变的 109 个基因,并确定了HSD17B4突变。由于覆盖率低,第二个HSD17B4突变最初并未在患者 2 中发现(假设仅存在 2/8 读数,其变异质量为 18.4,低于我们的阈值 20)。
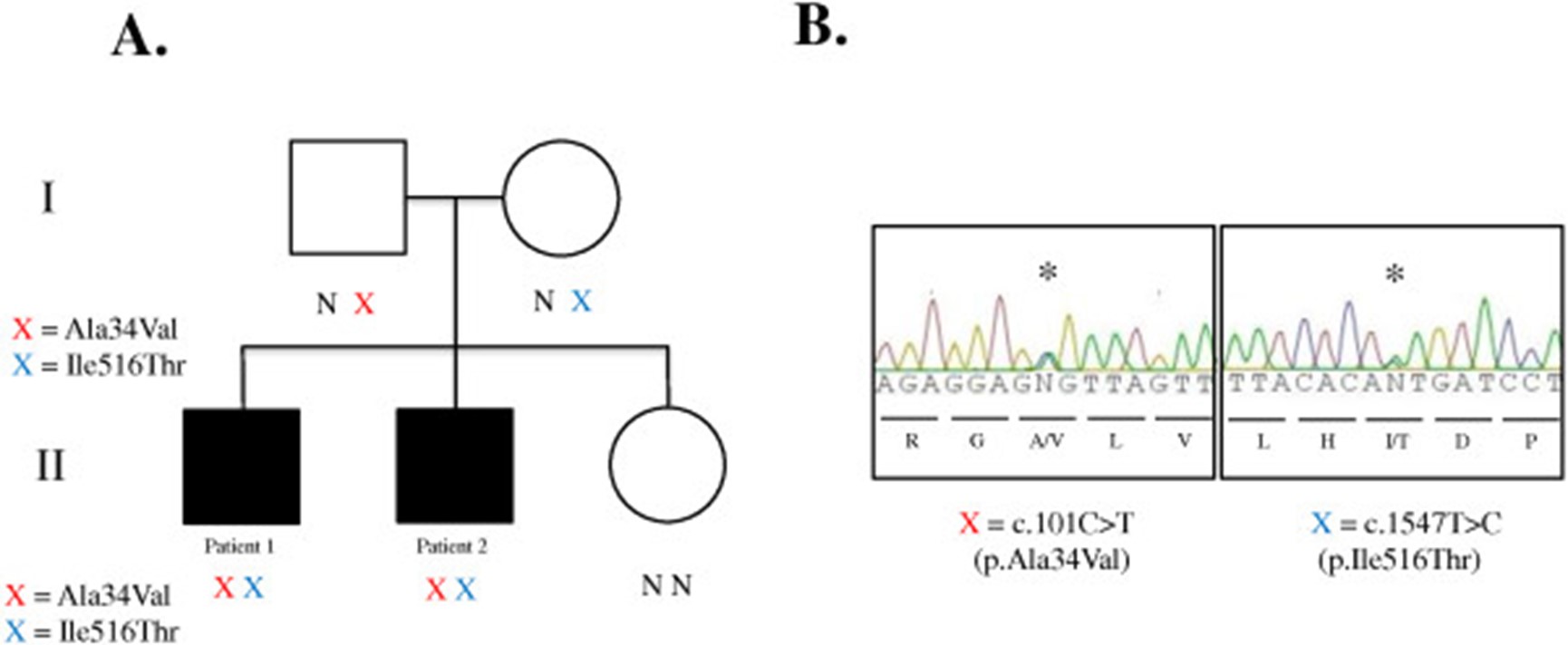
图 3:Sanger测序和分离。( A ) DBP 缺乏家系谱系。X=变体;N=正常。( B ) 外显子组测序鉴定的HSD17B4变体的Sanger 测序验证。使用外显子 2 和 18 侧翼的引物扩增基因组 DNA 用于测序(引物序列见正文)。星号表示杂合突变。红色 X = c.101C>T(p. Ala34Val)在 chr5:118792052。蓝色 X = c.1547T>C(p. Ile516Thr)在 chr5:118860954。
生化分析
血浆中 VLCFA 和支链脂肪酸(鹦鹉螺酸和植烷酸)的水平正常(表2). 血浆二十二碳六烯酸 (DHA) 水平也正常。使用快原子轰击电离质谱法分析尿液,未发现尿液胆汁酸分泌异常。遗传代谢疾病实验室的成纤维细胞研究显示正常的 VLCFA 水平和正常的 C26:0 β-氧化,但鹦鹉螺酸 β-氧化活性降低(表2). 过氧化氢酶免疫荧光研究显示过氧化物酶体染色在过氧化物酶体的数量和形态方面正常至接近正常(表2). 患者 1 中过氧化物酶体的大小略有增加。DBP 酶活性测量显示水合酶和脱氢酶活性降低(表2). 免疫印迹上 DBP 蛋白的量减少(表2)。
前额高腭弓高囟门增大人中长眼距过宽肌张力减退还有癫痫是不是遗传性DBP缺乏症的讨论
佳学基因对患有先前未确诊的神经退行性疾病的两个兄弟进行外显子组测序并进行基因解码基因检测,检测到 HSD17B4 基因中存在复合杂合突变 c.101C>T (p.Ala34Val) 和 c.1547T>C (p.Ile516Thr),这两个突变位点分别影响脱氢酶和水合酶结构域。这两种错义突变之前都有报道,但在这两种情况下,第二个错义突变都会影响另一个等位基因上的相同 DBP 结构域. 第一个案例是 p.Ala34Val 和 p.Phe237Ser 的复合杂合子;影响脱氢酶结构域的两个突变导致 III 型 DBP 和孤立的脱氢酶缺陷。第二种情况是 p.Ile516Thr 和 p.Asn457Tyr 的复合杂合子;在这种情况下,两种突变都发生在水合酶结构域内,导致 II 型 DBP 和孤立的水合酶缺陷。尽管这名 II 型 DBP 患者存活了 13.5 年以上,但认知和语言缺陷很明显。
对于在佳学基因检测的患者中观察到的相对较轻的临床表型,一个明显的解释是每个等位基因上只有一个结构域受到不太严重的突变的影响。临床表型显示毒性降低和生化测试表明正常的转录、翻译和 DBP 酶正常输入到完整的功能性过氧化物酶体中。后者得到了佳学基因检测患者中观察到的正常过氧化氢酶免疫荧光的支持,表明正常的过氧化物酶体生物发生和形态;在患有更严重的 DBP 缺乏症的患者中未发现这一情况. 关于 p.Ala34Val 突变的影响的信息很少,分析表明脱氢酶结构域的残留酶活性较低。p.Ile516Thr 位于水合酶亚基的二聚化界面,但并未完全消除二聚化,这意味着存在有残留的活性。残留水合酶活性(约正常下限的 40%)与免疫印迹上水合酶结构域缺失之间的明显差异可能部分是由于 L-双功能蛋白 (L-BP) 负责部分测量的水合酶活性,因为它可以将底物 THC:1-CoA 代谢为 (24 S ,25 S)-24OH-THC-CoA 的异构体,不能被 L-BP 的脱氢酶结构域进一步代谢。结果也可以部分地代表突变体 DBP 酶的结构改变和稳定性的组合。最后,两组同源二聚体(水合酶和脱氢酶结构域)可能彼此具有某种物理和功能关系。一个结构域中的突变有可能改变另一个单元的功能和稳定性,即使突变结构域(例如脱氢酶)与相邻的野生型结构域(例如水合酶)形成二聚体。因此,这种酶的体内功能似乎比体外研究预测的更复杂。
尽管由单一酶缺陷引起的其他一些过氧化物酶体疾病可能会出现在成年期(既酰基辅酶 A 氧化酶 (ACOX) 缺乏症和甾醇载体蛋白 X (SCPx) 缺乏症,但尚未报道 DBP 缺乏症。由于水合酶和脱氢酶活性均受到影响,佳学基因检测的患者在当前的 DBP 缺陷分类下将被视为 I 型。然而,绝大多数 I 型缺陷患者在编码截短或不稳定蛋白质的HSD17B4中有突变,导致严重的表型和较差的生存. 据佳学基因解码分析,患者的轻度临床和生化表型需要进行新的分类。因此,佳学基因致病基因鉴定基因解码提出了一种新的 DBP 缺陷变体,命名为 DBP IV 型,这是由于复合杂合突变影响了 DBP 的两个不同结构域,但与相对较温和的临床和生化表型相关。
《神经系统疾病的临床表征与基因序列变化》新提出的 DBP 缺乏亚型(IV 型)也适用于最近被诊断患有由HSD17B4 内的复合杂合突变引起的 Perrault 综合征的两姐妹,一个影响脱氢酶结构域,一个影响水合酶结构域,与佳学基因检测的患者相似。在这种情况下,姐妹们相对较轻的表型表现为感音神经性耳聋、轻度智力障碍、感觉运动性多发性神经病、身材矮小和卵巢发育不全。外显子组测序对于获得该诊断也是必不可少的,但未报告包括 DBP 酶活性测量在内的完整生化测试。佳学基因的患者与这些姐妹的不同之处在于她们正常的智力和青春期发育(在哥哥身上)。
过氧化物酶体疾病的总发病率约为每 5,000 个新生儿中有 1 个,这些病例中的大多数都很严重,因此很容易确定。与之前报道的患者形成鲜明对比的是,这里分享的两兄弟没有表现出任何新生儿或婴儿症状,而且他们继续表现出正常的认知。他们缓慢的 DBP 缺乏临床过程首次允许对 DBP 患者进行系列电生理诊断性测试;跨越数年的临床检查和神经传导研究(表 1) 记录反映小脑和感觉神经功能障碍增加的协调性逐渐下降。进行性感觉运动性多发性神经病表现出均匀的传导速度减慢,让人联想到许多遗传性脱髓鞘性多发性神经病(例如 Charcot-Marie-Tooth1 型)。年长的兄弟(患者 1)不仅表现出进行性脱髓鞘(延迟增加和传导速度减慢),而且还表现出进行性的、长度依赖性轴突损失的证据。将现成的外显子组测序引入罕见病诊所将导致识别出更多患有 DBP 缺陷较轻变异的患者,并将提高我们对这种疾病和其他疾病的表型谱和发病过程的理解。
