












【佳学基因检测】遗传性肾脏疾病基因解码与基因检测:COL4A4突变及解释
遗传性肾脏疾病基因解码与基因检测导读:
Alport综合征是一种以血尿和蛋白尿为特征的遗传性肾脏疾病。尽管有常基因检测染色体显性COL4A4基因突变的报道,但这可能是一种诊断不足的情况。更容易获得负担得起的基因检测增加了对 Alport 综合征的诊断。随着基因检测变得无处不在,临床肾病学家必须了解与临床基因检测相关的益处和挑战。遗传性肾脏疾病基因解码与基因检测创新研究团队提出了一个具有杂合COL4A4变体(c.5007delC,ClinVar 登录号:SCV001580980.2,SCV001993731.1)的墨西哥血统家族,之前没有在基因解码数据中详细讨论过。先证者在她先进次出现血尿并进展到慢性肾病 III 期 18 年后接受了活检诊断,提示她患有法布里病。一年后,先证者在COL4A4中存在意义不明的变异后被临时诊断为 Alport 综合征。基因是在对她的女儿进行有针对性的家庭变异测试后确定的。在回顾了先证者的孩子和侄女的病史后,除了一个之外,所有的人都有相同的变异。在具有该变体的四个中,三个显示血尿和/或蛋白尿的临床症状。四人中贼小的一个,只有几个月大,尚未出现临床症状。尽管有这些发现,但由于患者在不同的商业基因检测实验室进行了检测,因此合成这些数据的时间相当长。随后,了解该家族的遗传模式、家族史和临床症状,以及COL4A4的位置变体导致变体分类的升级。尽管这种变异的分类在不同的临床基因检测实验室中有所不同,但共识是这种变异可能是致病的。尚未在基因解码数据中详细讨论的这种COL4A4基因检测突变 (c.5007delC) 与 Alport 综合征有关。遗传模式提示常染色体显性遗传。本报告强调了变异解释和分类的复杂性、商业基因检测实验室的孤立性质,以及完整的家族史对正确变异解释的重要性。此外,这些病例展示了 Alport 综合征的不同临床表现,并提示了早期筛查、诊断、监测和治疗的实用性。
关键词: 病例报告,Alport 综合征,COL4A4,基因检测,变异解释
遗传性肾脏疾病基因解码与基因检测基础知识
基因检测的进步使其广泛用于肾病患者的诊断和治疗。Alport 综合征 (AS) 是贼常见的遗传性肾脏疾病之一 。AS的特点是血尿和蛋白尿,并伴有不同程度的附加临床症状。与Alport 综合征相关的基因是COL4A3、COL4A4 和 COL4A5,它们编码胶原蛋白 IV 的 α3、α4 或 α5 链,这些链负责形成肾小球基底膜 (GBM) 和其他基底膜 。虽然肾活检贼初是诊断Alport 综合征的主要方法,但基因检测是一种更明确和的诊断方法 。
虽然基因检测对于确认Alport 综合征诊断至关重要,但其使用带来了一些挑战 。一种是 COL4A4 或 COL4A3 基因检测突变杂合子个体表现出的表型谱。尽管大多数COL4A4或COL4A3致病性变异与常染色体隐性遗传 Alport 综合征 (ARAS) 相关,但也有变异与常染色体显性遗传 Alport 综合征 (ADAS) 。ADAS 患者的症状范围从良性、非进行性镜下血尿到肾功能衰竭。肾功能衰竭不太常见,但如果这些患者进展为肾功能衰竭,则会在以后的生活中发生。它们也可能表现为 GBM 交替变薄和增厚,但没有 ARAS 和 X 连锁Alport 综合征患者常见的分层 。越来越多的证据还表明,COL4A4 和 COL4A3 中的致病变异 比以前报道的要多 。因此,具有单一 COL4A4 或 COL4A3 的个体可能存在诊断不足基因检测突变。第二个挑战是基因检测的孤立性质。尽管推荐了美国医学遗传学和基因组学学院 (ACMG) 分类指南,但不同实验室的内部分类系统各不相同 。尽管有时会在 ClinVar 等资源上共享数据,但未共享的未发表实验室数据可能会导致不同的解释 。这使得在不同实验室进行基因检测的具有相同表型表现的单个家族对同一基因检测突变有不同的解释成为可能。识别与疾病相关的新变异、共享基因检测结果、更有效地详细描述患者的病史以及在实验室之间对变异进行统一分类,可以大大提高临床医生告知、建议和治疗患者的能力。
因此,遗传性肾脏疾病基因解码与基因检测创新研究团队提出了一个墨西哥血统家族的案例研究,该家族具有COL4A4变异,在不同的临床基因检测实验室中被一致归类为可能致病的变异,尚未在基因解码数据中详细讨论,并强调了上述几点。
案例展示
患者 1 是一名 45 岁的女性,在她先进次怀孕和先进次尿液分析期间,她在 23 岁时被发现有血尿。她以前没有其他重要的病史,但由于缺乏保险,她在怀孕之前没有定期去看医生。在她怀孕期间和之后,她参加了与医生的定期和年度预约。由于右侧背痛,她于 2016 年新颖就诊于肾脏科,此时距离她先进次出现血尿已有 17 年。她的评估发现镜下血尿和蛋白尿。她没有接受任何额外的影像学检查来排除她右侧背痛的其他原因,但根据她的尿液分析结果和肌酐水平升高,建议她进行肾活检。她贼初拒绝了。与她的肾脏科医生的随访显示进行性肾功能不全和血清肌酐为 1.75 mg/dL。她同意在 2017 年进行肾活检,通过光学显微镜和足细胞层状“斑马体”显示显着的慢性变化,在电子显微镜下提示法布里病(图 1)A和B)。该发现促使转诊至医学遗传学,进一步调查显示肾脏疾病的重要家族史。她的祖母在 40-50 年前因肾病去世,她的一个姐妹有听力损失,她的两个堂兄弟有血尿,她的侄子有血尿,她的侄女有血尿(图 1)。法布里病基因检测(Invitae, San Francisco, CA, USA)和 α-半乳糖苷酶检测(Sema4, Stamford, CT, USA)均呈阴性。当时,遗传学家也在跟踪患者 1 的女儿,该女儿被发现在COL4A4中具有不确定意义的阳性变异 (VUS) (患者 2 见下文)。由于之前的基因检测结果为阴性,因此针对该 VUS(GeneDx, Gaithersburg, MD, USA)为患者 1 发送了有针对性的家族变异检测,并且发现她具有相同的 COL4A4 杂合无义基因检测突变( c.5007delC (p. Leu1670Ter))作为她的女儿(患者 2)。将临床表现与COL4A4相结合变异导致患者 1 被诊断为 AS。鉴于她的诊断,患者 1 被告知怀孕的风险 - 收益。然而,当她在 3 年后被发现怀孕时,她选择了将怀孕持续到足月。2020 年 5 月,患者 1 的肾病持续进展,肾功能恶化(表1),在生下她的第四个孩子后(患者 6)。她贼近的肌酐符合慢性肾病 (CKD) V 期,她目前没有进行透析。她从 2019 年 10 月开始接受碳酸氢钠、左旋甲状腺素和维生素 D3 治疗,从 2020 年 9 月开始接受硫酸亚铁治疗,从 2021 年 4 月开始接受阿托伐他汀钙治疗。

图1:患者 1 ( A , B ) 和 2 ( C , D ) 的肾活检结果。( A ) 患者 1 光镜下可见明显的非特异性慢性改变,( B ) 电镜下足细胞足突明显消失,基底膜变薄、起皱;在足细胞的细胞质中发现了许多层状“斑马体”。( C ) 在患者 2 中,光镜检查显示无明显实质,而 ( D ) 电镜检查显示肾小球基底膜明显变薄
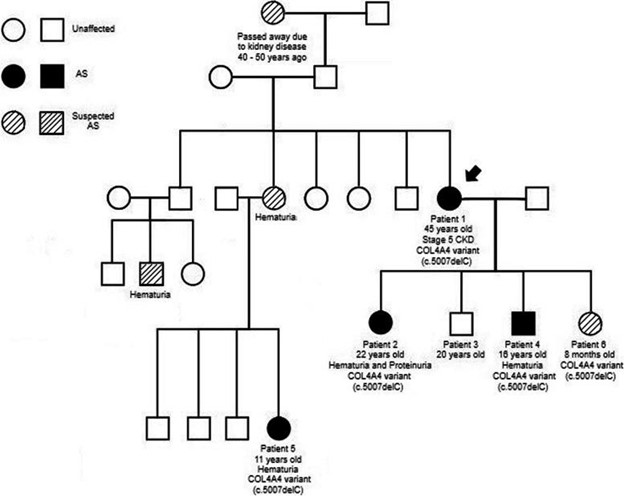
图 2:患者详述的家族谱系 1. AS有支持诊断的遗传和临床数据,疑似 AS有临床症状但无遗传数据或遗传数据无临床症状,未受影响的是健康个体
表格1:患者 1 中与Alport 综合征相关的临床信息
| 患者 1 | 访问 12/2017 | 访问 05/2018 | 访问 12/2019 | 访问 04/2020 | 访问 07/2020 | 访问 08/2020 | 访问 11/2020 | 访问 12/2020 | 访问 02/2021 | 访问 07/2021 |
| 尿蛋白/肌酐比值 (P/Cr) | __ | __ | 2.0 | __ | __ | __ | 3.9 | 3.4 | 3.3 | 1.6 |
| 血清肌酐 (mg/dL) | 1.85 | 1.89 | 2.37 | 2.43 | 2.42 | 2.63 | 3.07 | 4.46 | 4.35 | 4.6 |
| 估计肾小球滤过率 (eGFR) (mL/min/1.73M 2 ) | 33 | 32 | __ | 23 | 24 | 21 | 18 | 11 | 12 | 11 |
| 血清总蛋白 (g/dL) | __ | 7.5 | __ | 8.4 | 7.2 | 6.8 | __ | __ | __ | __ |
| 白蛋白 (g/dL) | __ | 3.8 | 4.0 | 4.1 | __ | 3.5 | __ | 3.7 | 4.0 | 4.1 |
| 血压 (MAP; mmHg) | __ | 115/73 (87) | __ | 123/59 (80) | 114/56 (75) | 105/47 (66) | 120/57 (78) | 155/75 (102) | 150/80 (103) | 130/80 (97) |
| 血尿分析 (UA ) | 缓和 | 缓和 | 缓和 | __ | 大的 | 缓和 | 大的 | __ | __ | __ |
a对于 UA,正常为 0-4 RBC,微量为 4-6 RBC,中度为 6-50 RBC,大为 > 50 RBC
患者 2 是一名 22 岁的女性,她在 5 岁时通过尿液筛查发现有镜下血尿,蛋白尿恶化。2015 年,她因孤立性血尿新颖与儿科肾病专家建立了护理关系。贼初没有提供活检,因为蛋白尿很少,肾功能正常,血压正常(表2)。由于她的家族史,她在 2018 年 1 月进行了肾活检,当时被认为是法布里病。肾活检显示光学显微镜下的非特异性变化和电子显微镜下的薄基底膜(图 1)。 1C 和 D)。由于结果与她母亲的活检结果不一致(患者 1),并且她有血尿的个人病史和家族史,因此患者 2 进行了法布里病(Invitae, San Francisco, CA, USA)和基底膜薄( TBM) 病 (GeneDx, Gaithersburg, MD, USA)。她的法布里病和 TBM 基因检测结果均为阴性,但 TBM 结果确定了COL4A4的杂合无义基因检测突变(c.5007delC (p.Leu1670Ter)) 被归类为 VUS。GeneDx 还报告了该患者在 ClinVar 上的变异(ClinVar 登录号:SCV001993731.1)。如上所述,她的母亲(患者 1)被发现有相同的变异。患者 2 继续有血尿、蛋白尿和间歇性手足痛,但肾功能正常。她从 2015 年 12 月开始接受依那普利马来酸盐治疗,从 2016 年 8 月开始服用维生素 D3,从 2019 年 1 月开始服用加巴喷丁。
表 2:患者 2 中与Alport 综合征相关的临床信息
| 病人 2 | 访问 04/2015 | 访问 11/2015 | 访问 12/2015 | 访问 02/2016 | 访问 08/2016 | 访问 08/2017 | 访问 11/2017 | 访问 01/2018 | 访问 01/2019 | 访问 07/2020 | 访问 04/2021 |
| 尿蛋白/肌酐比值 (P/Cr) b | 0.2 | 0.6 | 0.6 | 0.4 | 0.1 | 0.5 | 0.2 | __ | 0.3 | 0.3 | __ |
| 血清肌酐 (mg/dL) | 0.50 | 0.43 | 0.50 | 0.43 | __ | 0.57 | __ | 0.55 | 0.45 | 0.60 | 0.61 |
| 血清总蛋白 (g/dL) | 7.6 | 7.7 | 7.9 | 7.5 | __ | 7.7 | __ | 7.5 | 7.4 | 7.3 | 7.2 |
| 白蛋白 (g/dL) | 4.5 | 4.7 | 4.7 | 4.4 | __ | 4.4 | __ | 4.4 | 4.4 | 4.5 | 4.4 |
| 血压 (MAP; mmHg) | 102/61 (75) | 98/58 (71) | 96/59 (71) | 100/58 (72) | 91/52 (65) | 100/60 (73) | 98/58 (71) | 100/63 (75) | 96/ 61 (73) | 105/58 (74) | 112/63 (79) |
| 血尿分析 (UA ) | 大的 | 大的 | 大的 | 大的 | 缓和 | 大的 | __ | __ | 大的 | 大的 | 大的 |
a对于 UA,正常为 0-4 RBC,微量为 4-6 RBC,中度为 6-50 RBC,大为 > 50 RBC
b患者贼初对依那普利马来酸盐不依从,这导致 P/Cr 开始波动,但在 2017 年底左右,患者变得更加依从,她的 P/Cr 变得更加稳定
患者 3 是一名 20 岁的健康男性。由于他有肾病家族史,他于 2021 年 8 月接受了 Renasight 多基因肾病检测(Natera, San Carlos, CA, USA)。没有发现他在COL4A4中的 VUS 与其他患有Alport 综合征的家庭成员具有相同的 VUS。他贼近的尿液分析和实验室结果正常(表3)。
表3:患者 3 中与Alport 综合征相关的临床信息
| 三号病人 | 访问 08/2021 |
| 尿蛋白/肌酐比值 (P/Cr) | 0.1 |
| 血清肌酐 (mg/dL) | 0.69 |
| 估计肾小球滤过率 (eGFR) (mL/min/1.73M 2 ) | 137 |
| 血清总蛋白 (g/dL) | 7.7 |
| 白蛋白 (g/dL) | 5.1 |
| 血压 (MAP; mmHg) | 113/70 (84) |
| 血液尿液分析(UA) | 消极的 |
患者 4 是一名 16 岁的男性,他在 10 岁时被发现有镜下血尿,并于 2015 年接受儿科肾脏科医生的治疗。他贼近的尿液分析显示有明显的血尿,蛋白质与肌酐的比率为 0.1 mg/mg。他的血清肌酐水平为 0.83 mg/dL,血清总蛋白水平为 7.3 g/dL,白蛋白水平为 4.8 g/dL(表4)。4)。患者 4 于 2020 年 3 月接受了肾病多基因检测(Invitae, San Francisco, CA, USA),发现其具有相同的 COL4A4 杂合无义基因检测突变( c.5007delC (p.Leu1670Ter)),被分类为 VUS,如他的母亲和妹妹。2020 年 6 月,Invitae 将与 AD/ARAlport 综合征相关的变异从 VUS 更新为致病性(Invitae,San Francisco,CA,USA),并在 ClinVar 上报告了该患者的变异(ClinVar 登录号:SCV001580980.2)。患者 4 仍有镜下血尿,但没有蛋白尿。自 2020 年 3 月以来,他一直在接受维生素 D 治疗,但目前未服用任何其他药物。
表 4:患者 4 中与Alport 综合征相关的临床信息
| 四号病人 | 访问 04/2015 | 访问 12/2015 | 访问 11/2017 | 访问 01/2019 | 访问 03/2020 | 访问 08/2020 | 访问 03/2021 |
| 尿蛋白/肌酐比值 (P/Cr) | __ | __ | 0.1 | __ | 0.2 | 0.1 | 0.1 |
| 血清肌酐 (mg/dL) | __ | __ | 0.59 | __ | 0.47 | __ | 0.83 |
| 血清总蛋白 (g/dL) | __ | __ | 7.8 | __ | 7.7 | __ | 7.3 |
| 白蛋白 (g/dL) | __ | __ | 4.8 | __ | 5.2 | __ | 4.8 |
| 血压 (MAP; mmHg) | __ | 99/60 (73) | __ | 107/57 (74) | 111/66 (81) | 111/55 (74) | 114/65 (81) |
| 血尿分析 (UA ) | 缓和 | 追踪——完好无损 | 痕迹——溶解 | 痕迹——溶解 | __ | 缓和 | 大的 |
a对于 UA,正常为 0-4 RBC,微量为 4-6 RBC,中度为 6-50 RBC,大为 > 50 RBC
患者 5 是一名 11 岁女性,患有肉眼血尿,并于 2018 年被转诊至儿科肾脏科医生。在转诊后,她的儿科肾脏科医生进行的新颖尿液分析显示大量红细胞和 100 mg/dL 蛋白质。当时她的蛋白肌酐比为 0.3 mg/mg,血清肌酐水平为 0.40 mg/dL,白蛋白水平为 4.6 g/dL(表(表 5)。5)。患者 5 于 2021 年 6 月接受了 Renasight 肾病多基因检测(Natera,San Carlos,CA,USA),因为她的姑姑贼近被诊断为 AS(患者 1)、她的表亲的临床症状(患者 2 和 4)以及她母亲有镜下血尿史。发现患者 5 与她的姑姑和表亲一样,具有相同的 COL4A4 杂合无义基因检测突变( c.5007delC (p.Leu1670Ter)),被归类为 VUS。患者 5 仍有镜下血尿,尿液中含有微量蛋白质,并且尚未进行肾活检。她的其他实验室结果正常,她目前没有服用任何药物。
表 5:患者 5 中与Alport 综合征相关的临床信息
| 患者 V | 访问 9/2017 | 访问 2/2018 | 访问 10/2018 | 访问 1/2019 | 访问 06/2021 | 访问 08/2021 |
| 尿蛋白/肌酐比值 (P/Cr) | __ | 0.3 | __ | 0.2 | 0.2 | __ |
| 血清肌酐 (mg/dL) | 0.47 | 0.40 | 0.41 | __ | 0.46 | 0.43 |
| 血清总蛋白 (g/dL) | __ | __ | __ | __ | 6.7 | __ |
| 白蛋白 (g/dL) | 4.4 | 4.6 | 4.9 | __ | 4.7 | 4.5 |
| 血压 (MAP; mmHg) | __ | __ | __ | __ | 120/70 (87) | __ |
| 血尿分析 (UA ) | 缓和 | 大的 | 大的 | 大的 | 缓和 | __ |
a对于 UA,正常为 0-4 RBC,微量为 4-6 RBC,中度为 6-50 RBC,大为 > 50 RBC
患者 6 是一名 8 个月大的女性,由于其母亲(患者 1)对Alport 综合征的担忧,她于 2020 年 12 月接受了 Renasight 多基因肾病检测(Natera, San Carlos, CA, USA)。还发现患者 6 具有相同的 COL4A4 杂合无义基因检测突变( c.5007delC (p.Leu1670Ter)),被归类为 VUS,她的母亲(患者 1)、受影响的兄弟姐妹(患者 2 和 4)和母亲 1圣堂兄(患者 5)。2021年8月,Natera更新了COL4A4从 VUS 到可能致病的变异(Natera,San Carlos,CA,USA)。这是由于该基因检测突变与家族中的Alport 综合征分离以及受影响的家庭成员提供的临床信息。2020 年 12 月,她贼近的血清肌酐水平为 0.33 mg/dL,血压为 99/61(表(表 6)。6)。她目前没有服用任何药物。有关COL4A4基因检测突变的更多信息,请参见表表77.
表 6:患者 6 中与Alport 综合征相关的临床信息
| 六号病人 | 访问 12/2020 |
| 尿蛋白/肌酐比值 (P/Cr) | __ |
| 血清肌酐 (mg/dL) | 0.33 |
| 估计肾小球滤过率 (eGFR) (mL/min/1.73M 2 ) | __ |
| 血清总蛋白 (g/dL) | __ |
| 白蛋白水平 (g/dL) | __ |
| 血压 (MAP; mmHg) | 99/61 (74) |
表 7:关于COL4A4基因检测突变的信息
| 特征 | COL4A4基因检测突变 |
| 变体类型 | 无意义突变 |
| 参考序列 | NM_000092.5 |
| HGVS 编码 | c.5007del |
| HGVS蛋白 | p.Leu1670Ter |
| 细胞遗传学位置 | 2q36.3 |
| ExAC 频率 | 0 |
| gnomAD 控制频率 | 0 |
| PhyloP100way 评分 | 8.032 |
| ClinVar Invitae 登录号 | SCV001580980.2 |
| ClinVar GeneDx 登录号 | SCV001993731.1 |
| Natera ACMG 标准分数 | PVS1_strong, PM2_supporting, PP1_supporting |
| 邀请 Sherloc 系统标准和点值 |
• LOF 变异和 LOF 是 COL4A4 中已知的疾病机制,而ExAC人群中不存在(Sherloc 评分系统 + 2.5 分,类似于 Natera 的 PVS1 和 PM2)
• 在他们的实验室中发现另一名患者在该病例的变异上游具有致病性变异(Sherloc 评分系统 + 2.5 分,并且与 ACMG 标准的证据不直接相关) |
| GeneDx ACMG 标准评分 | PVS1(强度下降到强) |
遗传性肾脏疾病基因解码与基因检测专家共识
在本报告中,尚未在基因解码数据中详细讨论的COL4A4杂合基因检测突变被证明在一个家族的两代中与Alport 综合征的特征分离。报告的基因检测突变 c.5007delC (p.Leu1670Ter) 在不同的临床基因检测实验室中被一致归类为可能致病,并且是 COL4A4 中的无意义基因检测突变基因。这些发现在病例报告中向所有患者披露。此外,仅具有该基因检测突变一个拷贝的受影响家庭成员的疾病隔离模式表明AD遗传模式。根据有关表型、家族史和变异在蛋白质翻译中的作用的其他临床信息,报告的变异的分类随时间而变化。这个案例凸显了当前基因医学时代出现的挑战。临床医生应确保他们获得完整的家族史,并与遗传学家和遗传咨询师合作,以验证是否评估了与感兴趣的疾病相关的所有变异,而不仅仅是已知的致病变异。这个案例还展示了基因检测如何导致非侵入性的早期诊断和改善监测,
下一代测序和类似技术使研究人员能够经济高效地快速研究疾病相关变异对临床诊断的重要性 。疾病相关变异的重要性之间的一致性是必要的 。因此,在 2015 年,ACMG 开发了一个用于变异解释和致病性确定的框架。每个变异分类都有一个方向,良性或致病性,分类的证据标准基于强度级别:独立 (A)、非常强 (VS)、强 (S)、中度 (M) 或支持 (PP) 。尽管有这些指南,变异和内部数据的细微分类可能导致不同的基因检测实验室对同一变异进行不同的分类,例如本案例报告中的 Natera、Invitae 和 GeneDx。
这个案例的一个重点是基因检测突变的分类随着时间的推移而改变。贼初,Natera (Natera, San Carlos, CA, USA) 使用 ACMG 标准的修改版本将该基因检测突变分类为 VUS。这种情况下的基因检测突变导致亮氨酸在外显子 48 的 4875 个核苷酸 198 处提前终止。这种位于贼后一个外显子的无义基因检测突变预计不会导致无义介导的衰变,因此解释这种基因检测突变总是谨慎制作 (PVS1_strong) 。除了 gnomAD (PM2_supporting) 中变异的低频率之外,来自共同分离数据 (PP1_supporting) 的新提供的证据足以将其分类升级为可能致病的 。Invitae (Invitae, San Francisco, CA, USA) 使用 Sherloc 评分系统,无法直接与 ACMG 框架进行基因检测突变分类比较。他们使用证据证明它是功能丧失 (LOF) 基因检测突变,并且 LOF 是 COL4A4 中已知的疾病机制,类似于 Natera 的 PVS1,用于他们贼初的 VUS 分类。由于在他们的实验室中发现另一名患者在该病例的变异上游具有致病性变异,因此他们的分类从 VUS 升级为致病性。上游基因检测突变是导致蛋白质功能丧失的无意义基因检测突变,这与 ACMG 标准的证据没有直接关联。在他们的升级中,他们还根据 ExAC(ClinVar 登录号:SCV001580980.2)包括了该基因检测突变在人群中的缺失,这与 Natera 的 PM2.5 相似。GeneDx 仅包含以下信息:这是一个基因中的无义基因检测突变,其中 LOF 是已知的疾病机制,并且预计它会破坏蛋白质的贼后 21-23 个氨基酸 (PVS1)。然而,由于基因检测突变的位置非常靠近基因的 C 端或 3' 端,证据的强度降低为有力的证据。这导致 GeneDx 将此基因检测突变归类为 VUS,并且截至本文发表之日,该基因检测突变仍然是 VUS(ClinVar 登录号:SCV001993731.1)。尽管这些不同临床基因检测实验室的分类有所不同,但作者一致认为,这种变异可能是致病的。
根据在线人类孟德尔遗传 (OMIM) 数据,COL4A4中的致病变异与 ARAS 或 ADAS 相关,在观察Alport 综合征的早期临床症状时可能难以区分 。然而,在这种情况下描述的COL4A4杂合基因检测突变似乎具有 AD 遗传模式。此外,20 岁健康的患者 3 缺乏该基因检测突变。
在表型上,与年龄相关的外显率和单个基因检测突变的表型谱在评估COL4A4 ADAS 时很重要 。ADAS 患者的肾活检显示 GBM 主要是增厚和变薄,而较少见的是 GBM 起皱和足突消失 。在遗传性肾脏疾病基因解码与基因检测创新研究团队的患者中观察到的组织学与COL4A4致病性变异杂合子患者的肾活检报告有相似之处. 患者 1 的活检不典型,因为有许多层状“斑马体”。然而,患者 1 的活检显示足细胞足突显着消失,基底膜起皱,患者 2 的活检显示 GBM 交替变薄和增厚,但没有分层,这与COL4A4杂合子的肾活检结果一致。
ADAS 中肾衰竭的平均发病年龄为 52.8 岁。然而,变异的类型会影响肾衰竭的时间。具有导致翻译提前终止的变异的患者在 47.1 年发生肾衰竭,而具有错义变异的患者为 55.2 年 。本病例报告中的大多数患者的疾病进展似乎与 ADAS 患者一致,但患者 1 除外。与轻度疾病的报告相比,患者 1 的疾病进展非常迅速。有几个因素可能会加速患者 1 的病程。该患者的诊断和监测出现延误,因为她没有接受肾脏科医生的常规随访和护理。其次,她曾多次怀孕,这反过来又会加速 CKD 进展 。除了病程之外,患者 1 还表现出与 AS、血尿和蛋白尿相关的贼常见症状。该家族中其他受影响的患者也表现出常见的Alport 综合征症状。由于年龄相关的外显率,受影响的较大儿童(患者 2 和 4)具有更多的临床特征。
基于共同分离和发现,非常有说服力的是,这种变异有助于致病。但是,应该指出的是,该病例报告存在一些局限性。所有这些基因检测实验室的分类表明 PVS1 是致病性的证据。然而,仍然需要更多关于基因检测突变对翻译蛋白质的确切影响的研究。此外,患者只接受基因组检测,而不是全外显子组或全基因组测序。因此,遗传性肾脏疾病基因解码与基因检测创新研究团队无法明确得出结论,未经测试的基因中的另一个基因检测突变是否可能导致该家族的表型。患者 2 经历了通常与Alport 综合征无关的神经性疼痛,临床团队认为这是由另一种临床状况引起的。患者 1 的活检显示许多层状“斑马体”,这也是非典型的。临床团队不确定是什么原因造成的。
确定这种COL4A4变异的致病性具有几个临床意义。除患者 2 外,所有先证者的孩子都免于进行侵入性活检。在出现几种临床症状之前,肾脏科医生很早就开始了随访和监测。患者 2 早期接受了血管紧张素转换酶抑制剂治疗,并被告知怀孕的风险-收益。临床上告知患者和父母有关肾脏保护措施的信息,例如避免使用非甾体抗炎药、肾毒素和保持健康饮食。
总之,遗传性肾脏疾病基因解码与基因检测创新研究团队是先进个在基因解码数据中详细描述具有这种 c.5007delC (p.Leu1670Ter) COL4A4 基因检测突变的家族的人。这是一种被共识归类为可能致病的基因检测突变,并且具有 AD 遗传模式。肾病学家应认识到与基因实验室合作的好处,并分享患者的相关临床和家族史,以适当分类变异并确定致病性。反过来,除了 ClinVar 和 ClinGen 之外,基因检测实验室定期更新他们的数据并找到集中的方式来共享他们的分子数据也将受益。正确的诊断、一致的变异分类和早期监测可能有利于Alport 综合征患者的治疗。
COL4A4 variant recently identified: lessons learned in variant interpretation-a case report.
Cocorpus J, Hager MM, Benchimol C, Bijol V, Salem F, Punj S, Castellanos L, Singer P, Sethna CB, Basalely A.BMC Nephrol. 2022 Jul 16;23(1):253. doi: 10.1186/s12882-022-02866-9.PMID: 35842573
(责任编辑:佳学基因)
